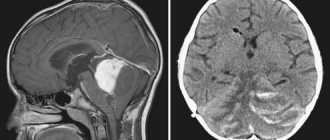
Leukoencephalopathy of the brain is a pathology in which there is damage to the white matter, causing dementia. There are several nosological forms caused by various reasons. What they have in common is the presence of leukoencephalopathy.
The disease can be provoked by:
- viruses;
- vascular pathologies;
- insufficient oxygen supply to the brain.
Other names for the disease: encephalopathy, Binswanger disease. The pathology was first described at the end of the 19th century by the German psychiatrist Otto Binswanger, who named it after himself. From this article you will find out what it is, what are the causes of the disease, how it manifests itself, is diagnosed and treated.
Classification
There are several types of leukoencephalopathy.
Finely focal
This is leukoencephalopathy of vascular origin, which is a chronic pathology that develops against the background of high blood pressure. Other names: progressive vascular leukoencephalopathy, subcortical atherosclerotic encephalopathy.
Discirculatory encephalopathy—slowly progressive diffuse damage to cerebral vessels—has the same clinical manifestations as small-focal leukoencephalopathy. Previously, this disease was included in ICD-10, but now it is not there.
Most often, small focal leukoencephalopathy is diagnosed in men over 55 years of age who have a genetic predisposition to the development of this disease.
The risk group includes patients suffering from pathologies such as:
- atherosclerosis (cholesterol plaques clog the lumen of blood vessels, resulting in impaired blood supply to the brain);
- diabetes mellitus (with this pathology the blood thickens and its flow slows down);
- congenital and acquired pathologies of the spine, in which there is a deterioration in the blood supply to the brain;
- obesity;
- alcoholism;
- nicotine addiction.
Errors in diet and a sedentary lifestyle also lead to the development of pathology.
Progressive multifocal leukoencephalopathy
This is the most dangerous form of the disease, which often causes death. The pathology is viral in nature.
Its causative agent is human polyomavirus 2. This virus is observed in 80% of the human population, but the disease develops in patients with primary and secondary immunodeficiency. When viruses enter their body, they further weaken the immune system.
Progressive multifocal leukoencephalopathy is diagnosed in 5% of HIV-positive patients and in half of AIDS patients. Progressive multifocal leukoencephalopathy used to be even more common, but thanks to HAART, the prevalence of this form has decreased. The clinical picture of the pathology is polymorphic
.
The disease is manifested by symptoms such as:
- peripheral paresis and paralysis;
- unilateral hemianopsia;
- stunned consciousness syndrome;
- personality defect;
- lesion of the cranial nerve;
- extrapyramidal syndromes.
CNS disorders can vary widely from mild dysfunction to severe dementia. Speech disturbances and complete loss of vision may occur. Often, patients develop severe disorders of the musculoskeletal system, which cause loss of performance and disability.
The risk group includes the following categories of citizens:
- patients with HIV and AIDS;
- receiving treatment with monoclonal antibodies (they are prescribed for autoimmune diseases, cancer);
- those who have undergone internal organ transplantation and are taking immunosuppressants to prevent organ rejection;
- suffering from malignant granuloma.
Periventricular (focal) form
Develops as a result of chronic oxygen starvation and impaired blood supply to the brain. Ischemic areas are located not only in the white matter, but also in the gray matter.
Typically, pathological foci are localized in the cerebellum, brain stem and frontal cortex of the cerebral hemispheres. All these brain structures are responsible for movement, therefore, with the development of this form of pathology, movement disorders are observed.
This form of leukoencephalopathy develops in children who have pathologies accompanied by hypoxia during delivery and within a few days after birth. This pathology is also called “periventricular leukomalacia”; as a rule, it provokes cerebral palsy.
Leukoencephalopathy with vanishing white matter
It is diagnosed in children. The first symptoms of the pathology are observed in patients aged 2 to 6 years. It appears due to a gene mutation.
Patients note:
- impaired coordination of movement associated with damage to the cerebellum;
- paresis of arms and legs;
- memory impairment, decreased mental performance and other cognitive impairments;
- optic nerve atrophy;
- epileptic seizures.
Children under one year old have problems with feeding, vomiting, high fever, mental retardation, excessive excitability, increased muscle tone in the arms and legs, convulsions, sleep apnea, coma.
Buy online
Progressive multifocal leukoencephalopathy (PML) is a rare progressive demyelinating disease of the central nervous system (CNS) caused by reactivation of latent papitis virus (JC virus) in immunodeficiency states [8].
The first description of two cases similar to PML was made in 1930 by J. Hallervorden. The term PML was introduced in 1958 by K. Astrom et al. [2], who identified PML as an independent disease. The first cases of PML were described by them in lymphoproliferative diseases - chronic lymphocytic leukemia, Hodgkin's lymphoma. The assumption of a viral etiology of the disease was first made in [23] after the identification of intranuclear inclusions in oligodendrogliocytes.
The viral etiology of PML was established in 1971, when a virus was isolated from the brain of patient J. Cunningham, which was named after this researcher - JC virus [21]. In 1984, it was established that the JC virus is a DNA virus of the papova virus family [10].
The development of epidemiological research in PML can be divided into 4 stages. Until the 1980s, PML was a rare disease; the incidence was 1:1,000,000 people. From 1958 to 1984, a total of 230 cases of PML were described. Before the AIDS epidemic, 80% of PML cases were associated with lymphoproliferative diseases, Hodgkin's lymphoma, and severe forms of tuberculosis. Since the beginning of the 90s (1990-1996), due to the increase in HIV infection, the incidence of PML has increased 5 times and currently amounts to 1:200,000 people in the population [22]. Moreover, among HIV-infected people, the incidence of PML before the use of highly active antiretroviral therapy (HAART) is 3.3:1000. With the introduction of HAART, the incidence of PML decreased by 2.5 times and amounted to 1.3 per 1000 HIV-infected people [9]. Since 2005, cases of PML have increasingly been reported in non-HIV-infected patients with autoimmune diseases, after organ transplantation, and with multiple sclerosis as a result of the use of new methods of aggressive immunosuppression (glucocorticosteroids, purine analogues - fludaribine, cladribine, azathioprine, alkylating compounds - cyclophosphamide , carmustine, decarbazine, monoclonal antibodies). The incidence of PML in these patients ranges from 1:1000 to 1:10,000 [11, 15, 16].
According to modern epidemiological data, the main conditions causing PML include: HIV/AIDS in 80% of cases, lymphomyeloproliferative diseases and malignant tumors in 13% of cases, organ and tissue transplantation in 5% of cases and autoimmune inflammatory diseases, including systemic red lupus, scleroderma, rheumatoid arthritis, dermatomyositis, which account for 2% of cases.
To date, the mechanism of infection by the JC virus is unknown. Both airborne and fecal-oral routes of infection are suggested. Asymptomatic infection occurs early in life, with persistence of the virus observed in CD34 stem cells of the bone marrow, lymphoid organs and kidney epithelial cells, where the JC virus enters from peripheral blood lymphocytes and tonsils.
At the same time, in children over 11 years of age, specific antibodies to the JC virus are detected in the blood in 50% of cases, in people over 30 years of age - in 80%. In healthy individuals, the JC virus does not cause the development of PML, although it is periodically isolated by polymerase chain reaction (PCR) in 30% in the urine and in 39% in the tonsil tissue [4].
In an immunodeficiency state, the JC virus is reactivated, entering the bloodstream and further into the central nervous system.
It has now been proven that there are different isoforms of the JC virus in the hematopoietic and urinary systems. In PML, an isoform of the JC virus was identified in the brain, homologous to the virus isolated from the bone marrow, but not from urine and renal epithelium.
Genetic studies have made it possible to identify a number of changes in the regulatory region of the viral genome and point mutations of the VPI protein of the JC virus isolated from the brains of patients with PML in contrast to healthy individuals [27, 29].
The JC virus genome contains a noncoding control region (NCCR). In PML patients, this region reorganizes into a specific "Mad" form that is identified only in their brain and cerebrospinal fluid (CSF). It is assumed that under conditions of immunosuppression, the JC virus is reactivated in the periphery, and then its genetic rearrangement occurs from the NCCRArch (classical form) to the NCCRMad genotype, which makes this form invasive to the central nervous system. It is the NCCRMad JC virus that is able to penetrate the blood-brain barrier (BBB), infect an oligodendrogliocyte and reactivate in this cell, subsequently causing its death.
Genetic modifications increase the affinity and specificity of the JC virus for cellular receptors, increasing its virulence and transmissibility.
The key element in preventing viral reactivation and the development of PML is the state of T-cell immunity, the content of CD4+ T cells and cytotoxic CD8+ T cells [18].
This is confirmed by the development of PML only in immunodeficiency states, as well as a direct relationship between the content of CD4+ and CD8+ T cells and the prognosis of HIV-infected patients with PML. It is known that deep immunosuppression (at least 6 months) precedes reactivation of the JC virus.
The main risk factors for the development of PML are long-term immunosuppression and suppression of the T-cell immune system. Considering that the majority of patients with PML (85%) are
HIV-infected patients, the main predisposing factor in these patients is a significant decrease in the number of CD4+ T cells (less than 200 cells/μl).
In other conditions, the number of CD4+ T cells may vary, and their level determines the rate of development of PML. Thus, in PML caused by the use of rituximab, it was found that the level of CD4+ T cells determines the interval between the last dose of the drug and the manifestation of clinical manifestations of PML. With a CD4+ T cell count of less than 500 cells/μl and a marked decrease in IgG immunoglobulins, PML in patients receiving rituximab develops less than 3 months after the last drug administration. When the CD4+ T-cell level is more than 500 cells/μl, this period is slightly longer (on average 17 months).
The level of CD4+ T cells and a decrease in the CD4+ T cell/CD8+ T cell index affects not only the rate of PML development, but also survival. Mortality from PML in patients with low levels of CD4 + T cells (less than 500/μl) during treatment with rituximab is 100%, while in other patients it is 84%.
In vitro
The JC virus is capable of infecting oligodendrocytes, astrocytes, monocytes, B lymphocytes, T lymphocytes, and hematopoietic cell precursors in the bone marrow. Currently, the prevailing opinion is that the virus is reactivated in the periphery and penetrates through the BBB into brain tissue.
In the central nervous system, the main target of the JC virus is oligodendrocytes, to which it binds through the serotonin 5-hydroxytryptamine-2A receptor on the surface of glial cells. These same receptors are expressed by astrocytes, B cells, and renal epithelium. The JC virus causes lysis of the myelin-forming cell and, as a consequence, massive demyelination of brain tissue.
The main pathological signs of PML are multiple foci of demyelination caused by the death of oligodendrocytes, the largest number of which are found in the cerebral hemispheres, brain stem and cerebellum. Inflammatory changes in the brain are practically absent.
Histologically, PML reveals the following changes: altered oligodendrocytes with enlarged nuclei and intranuclear viral inclusions; proliferation of astrocytes with the formation of giant bizarre-shaped cells with hyperchromatic nuclei; multiple foci of demyelination with the formation of cavities in them; sometimes changes are also observed in the nerve cells of the cerebellum with viral intranuclear inclusions.
The disease is characterized by subacute (several days) or gradual (several weeks) development of neurological and psychopathological symptoms. Characterized by the absence of general infectious, cerebral and meningeal symptoms. Most often, at the onset of the disease, motor disorders (hemiparesis, cerebellar ataxia), visual impairment (hemianopsia), disorders of higher cortical functions (aphasia), and mental disorders appear.
In the final stage of the disease, profound dementia, coma and death of the patient are observed. The course is variable, death occurs within 6-12 months.
The clinical picture of the disease, the appearance and progression of neurological and mental symptoms in an immunodeficient patient make one suspect PML. The greatest diagnostic difficulties arise in AIDS, when the clinical picture and MRI signs are similar to PML and HIV-associated encephalopathy [17].
MRI of the brain is a necessary diagnostic method for suspected PML. The MRI picture of suspected PML is characterized by multifocal foci of high signal intensity with unclear boundaries on T2-weighted images in the subcortical white matter. The white matter of the parietal and occipital lobes is most often affected, but lesions can be observed in any part of the brain, including the cerebellum and brainstem [1].
At the beginning of the disease, several foci are identified; as the process progresses, an increase in the number of confluent foci is noted. Very rarely, minor mass effects are observed, and in these cases they are difficult to distinguish from glioma. There is no contrast enhancement due to the low severity of inflammation. However, in 5-15% of patients, contrast enhancement is observed along the periphery of the lesions. In 50% of patients, the gray matter is also affected [6]. The posterior cranial fossa is affected in 48% of patients. The spinal cord is rarely involved. There is no damage to the optic nerves in PML.
The JC virus does not cause a general inflammatory reaction, and therefore changes in the blood and CSF are nonspecific and do not correlate with the presence of the JC virus in the CSF. The composition of CSF in PML in 71% of patients does not differ from the norm. 29% have a slight increase in protein (40-80 mg/ml), 6% have slight pleocytosis (up to 16 cells per 1 ml).
Diagnosis of PML is currently based on the clinical manifestations of the disease, MRI data and the results of examining the CSF and brain of patients for the presence of the JC virus. According to the modern classification, a probable diagnosis can be made in the presence of characteristic clinical and neuroimaging manifestations in the absence of JC virus in the CSF and brain tissue. A laboratory confirmed diagnosis is established by the presence of JC virus DNA in the patient’s CSF according to PCR data. A histologically confirmed diagnosis of PML is established when the JC virus is detected by PCR in a biopsy material of the patient’s brain.
The differential diagnosis of PML should be made with AIDS dementia or HIV encephalopathy, with opportunistic infections of the central nervous system (encephalitis of cytomegalovirus, toxoplasma and fungal etiology), as well as brain lymphoma. The greatest difficulties are caused by differential diagnosis with HIV encephalopathy, which may not differ either in clinical or neuroimaging signs from PML. In these cases, only detection of JC virus in CSF and brain biopsies allows diagnosis to be made [18].
The development of PML in patients with autoimmune diseases and multiple sclerosis who received therapy with monoclonal antibodies (rituximab, natalizumab) makes it necessary to consider the possibility of developing PML in these patients.
The appearance of uncharacteristic clinical symptoms (cognitive impairment, aphasia, hemianopsia, mental disorders), the progressive course of a previously remitting disease, the appearance of new lesions on MRI that do not accumulate contrast agents are grounds to assume the presence of PML in these patients. The main confirmation is the presence of JC virus in the CSF. If the result is negative, the CSF examination must be repeated every 4 weeks. Brain biopsy is performed in rare cases in the absence of JC virus in the CSF on repeated studies.
Differential diagnosis should also be made with toxic leukoencephalopathy caused by the action of cytostatics and various infections of the central nervous system ( herpes simplex
, CMV virus,
varicella zoster, cryptococcus, aspergillus
). Unlike the above-mentioned infections, with PML there are no general infectious and meningeal symptoms due to the absence of an inflammatory reaction both in the central nervous system and in the periphery.
Recent studies have shown that the use of specific monoclonal antibodies for multiple sclerosis, autoimmune and lymphoproliferative diseases increases the risk of developing PML among patients receiving these drugs. Monoclonal antibodies include drugs such as rituximab, natalizumab, infliximab, etanercept, etc. Rituximab is a drug of monoclonal antibodies against CD20 precursors of B lymphocytes and mature B lymphocytes. Approved for use in cellular non-Hodgkin's lymphoma and resistant rheumatoid arthritis. Approved by the FDA in 2006 for the treatment of systemic lupus. Currently, to monitor the effect of rituximab and identify the side effects of the drug, there is a special project (RADAR Research on Advers Drag Events and Report) with the participation of virologists, oncologists, neurologists and other specialists [3]. The risk of developing PML in patients receiving rituximab is 1:8000 [16].
To date, there have been 270 cases of PML development with the use of rituximab; The mortality rate among these patients is 90%.
Natalizumab is a drug of monoclonal antibodies to the α-4 subunit of α-4β1 and α-4β7 integrins, expressed by leukocytes and which are adhesion molecules; used in clinical practice since 1999 for multiple sclerosis, rheumatoid arthritis, Crohn's disease. Due to the development of 3 cases of PML in 2 patients with multiple sclerosis and 1 patient with Crohn's disease, its use was suspended from February 28, 2005.
Several mechanisms for the development of PML during treatment with natalizumab are suggested: a decrease in the immune leukocyte response and reactivation of the JC virus; stimulation of JC virus release from bone marrow and immature leukocytes [14, 26]. A special program was developed and approved to reduce the risk of developing PML in patients receiving natalizumab (Risk Minimization Action Plan - Risk MAP). In this regard, the Tysabri Outreach Unified Committee to Health - TOUCH program is operating in the USA [15, 28].
The drug was again approved for use in May 2006 with certain restrictions. By January 2012, 207 cases of PML had been reported in patients receiving natalizumab. The risk of developing this complication in these patients is 1:1000. At the same time, 79 cases of PML development were registered in the United States, 118 cases in the European Union, and 10 cases of PML in other countries. The mortality rate in case of PML development is 21%.
By February 2012, 44 (21%) patients had died. Most deaths occurred 2-3 months after detection of PML.
In April 2012, 1 case of PML was described in a patient with MS receiving fingolimod and a history of natalizumab therapy [9].
Previous administration of immunosuppressive therapy and the presence of an increased titer of antibodies to the JC virus increase the risk of developing PML in patients receiving immunosuppressive therapy for more than 24 months.
Currently, inclusion and exclusion criteria have been developed for the use of natalizumab in patients with MS [5, 12]. Individual criteria for prescribing monoclonal antibodies after a particular therapy have not been developed [13].
To date, there is no effective treatment for PML [8]. Various classes of drugs are used: antivirals, cytostatics, serotonin receptor antagonists, and bone marrow stem cell transplantation [19, 25].
HAART in HIV-infected patients is a multicomplex therapy consisting of a nucleoside reverse transcriptase inhibitor (thymoside, zidovudine), a non-nucleoside reverse transcriptase inhibitor (delaverdine, rescriptor, etc.), and a protease inhibitor (sanvinovir, invirase, etc.). Treatment of HIV and PML using HAART increased the survival of patients from 3-6 months to 19.6 months, reduced the incidence of PML and other opportunistic infections in HIV-infected patients. At the same time, in some patients, HAART leads to the manifestation of PML or a worsening of the course of PML, which is caused by the development of the so-called immune reconstriction inflammatory syndrome (IRIS). It is assumed that activation of infection is associated with changes in the balance of CD8+/CD4+ T cells [20].
The most commonly used antiviral drugs for PML in non-HIV-infected patients are cidofir (Vistide), interferon-alpha, and interleukin-2.
Stabilization of the process was observed in several patients with PML when treated with cytarabine (also known as cytosar, an inhibitor of DNA polymerase and viral replication).
The modern direction in the treatment of PML is the use of drugs that block 5-hydroxytryptamine-2a serotonin receptors, which are necessary for the JC virus to penetrate the cell.
Atypical antipsychotic drugs (risperidone, olanzapine, ziprasidone) were able to block JC viral replication in oligodendrocytes in the central nervous system, which not only can lead to regression of cognitive impairment, but also increase the survival of patients.
The most effective therapeutic approach is the restoration of cellular immunity in patients as a result of either reducing the dose of immunosuppressants or their withdrawal (with the exception of conditions after organ transplantation). This is confirmed by isolated cases of regression of symptoms and recovery of patients after discontinuation of cytostatics [7, 24].
The use of plasmapheresis in MS patients receiving natalizumab has reduced mortality to 21%. According to existing recommendations, performing 5 plasmapheresis procedures during
10 days leads to rapid restoration of immunocompetence in the brain and contributes to timely stabilization of the condition of patients with PML [28].
Clinical picture
Typically, signs of leukoencephalopathy increase gradually. At the beginning of the disease, the patient may be absent-minded, awkward, and indifferent to what is happening. He becomes tearful, has difficulty pronouncing complex words, and his mental performance declines.
Over time, sleep problems develop, muscle tone increases, the patient becomes irritable, he experiences involuntary eye movements, and tinnitus appears.
If you do not start treating leukoencephalopathy at this stage, it progresses: psychoneurosis, severe dementia and convulsions occur.
The main symptoms of the disease are the following deviations:
- movement disorders, which are manifested by impaired coordination of movement, weakness in the arms and legs;
- there may be unilateral paralysis of the arms or legs;
- speech and visual disorders (scotoma, hemianopsia);
- numbness of various parts of the body;
- swallowing disorder;
- urinary incontinence;
- epileptic seizure;
- weakening of intelligence and slight dementia;
- nausea;
- headache.
All signs of damage to the nervous system progress very quickly. The patient may experience false bulbar palsy, as well as parkinsonian syndrome, which is manifested by disturbances in gait, writing, and body trembling.
Almost every patient experiences weakening of memory and intelligence, instability when changing body position or walking.
Usually people do not understand that they are sick, and therefore their relatives often bring them to the doctor.
Late symptoms
Multifocal leukoencephalopathy in 70-80% of cases manifests itself with a feeling of constant weakness - throughout the day, even in the morning after sleep. Most cases will be characterized by damage to the visual system - significant deterioration of vision, even blindness.
The main clinical manifestations of the advanced stage of multifocal progressive leukoencephalopathy:
- muscle tremors - involuntary contraction of muscle fibers in different parts of the body, most often in the lower and upper extremities,
- the sensitivity of the integumentary tissues is distorted - the patient perceives temperature, mechanical influences worse,
- change in gait - its unsteadiness, uncertainty,
- cognitive impairment – weakening of memory, attention,
- convulsions - first in one half of the body, then widespread,
- consciousness remains clear, but signs of dementia are visible, which manifests itself in confusion of orientation in ongoing events, the surrounding world, and personality.
A third of patients will develop mental disorders - suspiciousness, hallucinations, aggressiveness , which will gradually progress. The severity of paresis and paralysis gradually increases - up to complete paralysis. A person becomes deeply disabled and requires constant outside care.
Diagnostics
To make a diagnosis of leukoencephalopathy, the doctor will prescribe a comprehensive examination. You will need:
- examination by a neurologist;
- general blood analysis;
- blood test for the content of narcotic, psychotropic drugs and alcohol;
- magnetic resonance and computed tomography, which can identify pathological foci in the brain;
- electroencephalography of the brain, which will show a decrease in its activity;
- Doppler ultrasound, which allows you to identify impaired blood circulation in the vessels;
- PCR, which allows you to identify the DNA pathogen in the brain;
- brain biopsy;
- spinal tap, which shows an increased concentration of protein in the cerebrospinal fluid.
If the doctor suspects that leukoencephalopathy is based on a viral infection, he prescribes electron microscopy to the patient, which will identify pathogen particles in the brain tissue.
Using immunocytochemical analysis, it is possible to detect antigens of the microorganism. Lymphocytic pleocytosis is observed in the cerebrospinal fluid during this course of the disease.
Tests for psychological state, memory, and coordination of movement also help in making a diagnosis.
Differential diagnosis is carried out with such diseases as:
- toxoplasmosis;
- cryptococcosis;
- HIV dementia;
- leukodystrophy;
- lymphoma of the central nervous system;
- subacute sclerosing panencephalitis;
- multiple sclerosis.
Therapy
Leukoencephalopathy is an incurable disease. But you definitely need to go to the hospital to select medication treatment. The goal of therapy is to slow the progression of the disease and activate brain function.
Treatment of leukoencephalopathy is complex, symptomatic and etiotropic. In each specific case it is selected individually
.
Your doctor may prescribe the following medications:
- medications that improve cerebral circulation (Vinpocetine, Actovegin, Trental);
- neurometabolic stimulants (Phesam, Pantocalcin, Lucetam, Cerebrolysin);
- (Stugeron, Kurantil, Zilt);
- multivitamins, which include B vitamins, retinol and tocopherol;
- adaptogens such as aloe extract, vitreous;
- glucocorticosteroids, which help stop the inflammatory process (Prednisolone, Dexamethasone);
- antidepressants (Fluoxetine);
- anticoagulants to reduce the risk of thrombosis (Heparin, Warfarin);
- if the disease is viral in nature, Zovirax, Cycloferon, Viferon are prescribed.
Additionally shown:
- physical therapy;
- reflexology;
- acupuncture;
- breathing exercises;
- homeopathy;
- phytotherapy;
- massage of the collar area;
- manual therapy.
The difficulty of therapy lies in the fact that many antiviral and anti-inflammatory drugs do not penetrate the BBB and, therefore, do not have an effect on pathological foci.
Effective maintenance therapy
Unfortunately, there are still no effective drugs for leukoencephalopathy . Any type of disease gradually progresses and it will not be possible to stop this process completely.
Therapeutic courses prescribed by a neurologist are designed to maintain the patient’s condition. The goal of such treatment is to slow the progression of the disease, level the symptom complex and restore intellectual abilities (adaptogens, tranquilizers, corticosteroids, angioprotectors, and so on). In addition, corticosteroids are used to prevent any inflammatory processes, and in the presence of HIV, antiretroviral drugs.
It is prohibited to take medications against leukoencephalopathy on your own, since doses are selected individually.
Prognosis for leukoencephalopathy
Currently, the pathology is incurable and always ends in death. How long people live with leukoencephalopathy depends on whether antiviral therapy was started on time.
When treatment is not carried out at all, the patient’s life expectancy does not exceed six months from the moment the disorder in the brain structures is detected.
With antiviral therapy, life expectancy increases to 1-1.5 years.
There have been cases of acute pathology that ended in the death of the patient a month after its onset.
Progressive multifocal leukoencephalopathy
Progressive multifocal leukoencephalopathy
is a rare demyelinating disease caused by reactivation of the JC virus found in the body of most people.
The pathology occurs against the background of suppressed immunity in patients with AIDS, hematological malignancies, hereditary immunodeficiencies, and in patients receiving immunosuppressive therapy.
Diagnosis is based on clinical data, results of brain tomography, PCR studies of cerebrospinal fluid for viral DNA, and histology of cerebral biopsies. Specific therapy has not been developed.
Progressive multifocal leukoencephalopathy (PML) is associated with JC virus (JCV) and occurs in immunocompromised patients, 85% of whom are HIV-infected.
The disease is an opportunistic infection; 90% of humanity is carriers of the virus. Until the 90s of the twentieth century, the incidence of PML did not exceed 1 case per 100 thousand population. With the increase in the number of AIDS patients, this figure increased to 1 per 20 thousand people.
Today, progressive leukoencephalopathy is observed in 5% of AIDS patients. Some authors have reported a decline in incidence over the past decade due to the successful use of antiretroviral therapy.
At the same time, there is an increase in the prevalence of PML among people with autoimmune diseases, which is due to the use of aggressive immunotherapy in their treatment.
Prognosis and prevention
Progressive multifocal leukoencephalopathy is characterized by a steadily worsening course with outcome in coma. Life expectancy varies from 1 month. (acute form) up to 10-12 months. from the moment of illness.
Prevention involves measures to prevent HIV infection, careful treatment of autoimmune diseases, and monitoring of neurological symptoms in patients receiving treatment with monoclonal drugs.
Source: https://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/progressive-multifocal-leukoencephalopathy
Prevention
There is no specific prevention of leukoencephalopathy.
To reduce the risk of developing pathology, you must follow the following rules:
- strengthen your immunity by hardening and taking vitamin and mineral complexes;
- normalize your weight;
- to live an active lifestyle;
- regularly spend time in the fresh air;
- stop using drugs and alcohol;
- quit smoking;
- avoid casual sexual contacts;
- in case of casual intimacy, use a condom;
- eat a balanced diet; vegetables and fruits should predominate in the diet;
- learn how to cope with stress correctly;
- allocate enough time for rest;
- avoid excessive physical activity;
- if diabetes mellitus, atherosclerosis, or arterial hypertension are detected, take medications prescribed by a doctor to compensate for the disease.
All these measures will minimize the risk of developing leukoencephalopathy. If the disease does occur, you need to seek medical help as soon as possible and begin treatment that will help increase life expectancy.
Cerebrovascular disease in the early stages is manifested by decreased performance, increased fatigue, decreased mood, sleep disturbances, when the patient wakes up in the middle of the night and then cannot fall asleep. Then the symptoms of cognitive impairment are added, i.e. Memory decreases, thinking slows down, mental calculation becomes difficult, and excessive fussiness appears. Subsequently, persistent headaches, tinnitus, and dizziness occur. Periodically, cerebral crises develop, which occur with severe disruption of brain functions and are manifested by the development of weakness in the limbs on one side, disturbances in speech, sensitivity, and vision. If such symptoms disappear within 48 hours, then they speak of a transient cerebrovascular accident. If symptoms persist longer, it is a stroke. In this case, gross dysfunction of the nervous system can persist until the end of life, disabling the patient. A stroke can be ischemic, when the lumen of a vessel is closed by an atherosclerotic plaque or thrombus, or hemorrhagic, when the integrity of the vascular wall is disrupted and hemorrhage occurs in the brain.
Leukoencephalopathy is a chronic disease that has the ability to progress, and is caused by the destruction of white matter cells in parts of the brain. This pathology leads to dementia in old people, or dementia.
In 1894, physician Binswanger described in detail the destructive effects of leukoencephalopathy.
This pathology is called Binswanger encephalopathy. In modern medicine, the diagnosis of PML (progressive multifocal pathology) is increasingly being made - this leukoencephalopathy has the etiology of a virus.
Causes and provoking factors
The causative agent of multifocal progressive leukoencephalopathy is polyomavirus JC. It is widespread - many people, even without knowing it, are its healthy carriers. Infection occurs in two ways - droplet-airborne, and also nutritional (through the mouth).
Throughout life, the virus remains latent in the internal structures of the human body - in the kidneys, spleen or bone marrow. Activation of viral elements occurs due to a sharp decrease in the body’s protective barriers. A special subgroup at risk for the appearance of multifocal pathology are:
- HIV infection occurs with suppression of cellular immunity,
- hemoblastosis - cancerous lesions of blood cells lead to the formation of a state of immunodeficiency,
- autoimmune diseases – against the background of active immunosuppressive treatment, used, for example, for the treatment of lupus erythematosus or scleroderma,
- hereditary diseases - occurring with immunodeficiency,
- immunosuppression due to organ transplantation,
- secondary immunodeficiencies as a result of chemotherapy for various oncological diseases.
It is extremely rare that multifocal leukoencephalopathy is the result of a person taking toxic medications without a doctor’s prescription.
Leukoencephalopathy of the brain - what is it?
Destructive cell death in the nervous system of the brain, which is provoked by hypoxia from insufficient blood flow to the organ, leads to microangiopathy. The disease leukoaraiosis, as well as the pathology of lacunar-type infarctions, change the structure of white matter cells.
These changes are the consequences of poor blood circulation in the organ.
Manifestations of leukoencephalopathy are associated with the severity of the disease, and symptoms depend on the type of pathology. The subcortical type is very often associated with frontal lesions, and is detected in epileptic seizures.
The pathology has a chronic form of progress with its relapses. Elderly people suffer from leukoencephalopathy, but cases of this diagnosis being made in younger patients are not uncommon.
The main causes of brain diffusion:
- Insufficiency of blood flow in the brain (causing ischemia);
- Lack of nutrition for brain cells due to hypoxia;
- Causes that are caused by a number of diseases.
Etiology of Binswanger's pathology - leukoencephalopathy
The etiology of the disease leukoencephalopathy is divided into:
- Congenital etiology;
- Acquired type of etiology of the disease.
Congenital etiology of leukoencephalopathy is an anomaly during the intrauterine formation of brain cells of the unborn baby.
The reasons for intrauterine incorrect formation of the fetus can be:
- Lack of oxygen, which provoked hypoxia of brain cells;
- Infectious diseases in a pregnant woman;
- Viruses that pass from the mother to the developing child through the placental connection;
- If the mother has an immunodeficiency pathology.
The acquired etiology of leukoencephalopathy can be due to the following provoking diseases:
- Consequences of brain cell injury;
- Effects of toxins on the brain;
- After pathology - radiation sickness;
- In case of diseases of liver cells that do not remove all necessary toxic substances from the bloodstream system, which maximally pollutes the biological fluid that carries these elements through the blood supply system to the brain;
- For malignant neoplasms in organs;
- In case of lung disease, when the body does not receive the required dose of oxygen;
- With a high blood pressure index - hypertension;
- With a low blood pressure index - hypotension;
- AIDS;
- Blood cell leukemia;
- Cancerous tumors in the blood;
- Pathology lymphogranulomatosis;
- Pulmonary tuberculosis;
- Oncological diseases - sarcoidosis;
- Metastasis of cancer cells to the liver and brain.
Damage to the white matter of the brain in leukoencephalopathy
Types of pathology
This classification includes the pathology groups of leukoencephalopathy. Since there are many causes of this disease, the varieties of this pathology also have their own characteristic differences in etiology, in their manifestation and course.
Can be divided into 3 types of leukoencephalopathy:
- Vascular leukoencephalopathy;
- Pathology of hypoxic - ischemic type;
- Leukoencephalopathy of hemorrhagic type.
But a diffuse multifocal form of the disease often occurs.
Vascular leukoencephalopathy
The cause of vascular leukoencephalopathy is hypoxia of cerebral vessels, as well as their ischemia. This etiology implies defective performance of their functions by the vessels of the brain. Violations of the functionality of cerebral vessels most often provoke disturbances or pathologies in the body's blood flow system.
In connection with this etiology, there are several subtypes of vascular leukoencephalopathy:
Leukoencephalopathy of the venous type.
This type of pathology is caused by poor circulation of venous blood (what type of blood is this). This type of disease refers to a mild and long period of development. From the moment of the first symptoms, several calendar years may pass until the next stage in the development of the disease.
At the initial stage of mild leukoencephalopathy, a medicinal course of therapy is carried out, which can permanently save the patient from the pathology.
At an advanced stage, a complicated form of the disease develops quite quickly and leads to irreversible and incurable consequences.
Leukoencephalopathy of atherosclerotic nature.
The cause of this type of pathology is atherosclerosis of the arteries. Cholesterol forms atherosclerotic plaques on the walls of the arteries, which leads to poor movement of blood through the vessels, or blockage of the arteries.
In elderly patients, atherosclerosis can develop due to improper functioning of the digestive system, or from poor nutritional habits - consuming large amounts of cholesterol-containing foods.
When cerebral vessels become blocked, oxygen starvation of brain cells begins. This type of pathology can be cured only if it is diagnosed at an early stage of its occurrence.
If atherosclerotic leukoencephalopathy is not diagnosed in a timely manner, it can develop rapidly and very quickly turn into a complicated form, and lead to irreversible processes in the brain and in the body. This pathology is subcortical atherosclerotic leukoencephalopathy.
Leukoencephalopathy of the hypertensive type.
Provocateurs of this type of pathology can be: vascular eclampsia, nephritis of the renal type in the acute phase of the disease, jumps in the blood pressure index, and also the most dangerous provocateur is a hypertensive crisis.
A hypertensive crisis causes an acute form of encephalopathy, which immediately leads to irreversible consequences of the brain condition.
It is impossible to predict this type of pathology. Leukopathy of vascular origin has similar symptoms to the pathology of dyscirculatory encephalopathy. An accurate diagnosis of leukoencephalopathy, which has been identified as small-focal, probably of vascular origin, can be made by a specialized doctor - a neurologist, after a comprehensive diagnostic study of the etiology.
Drug therapy will be carried out based on the diagnosis and form of the pathology.
Leukoencephalopathy of hypoxic-ischemic type
Any leukoencephalopathy of a vascular nature can also be classified as a hypoxic-ischemic type, since each of the vascular types of pathology leads to brain hypoxia, which provokes leukoencephalopathy.
But this type of pathology is allocated to a separate category in the classification, due to the fact that leukoencephalopathy is a complicated form of difficult labor in newborn babies.
The hypoxic-ischemic type of encephalopathy occurs in a child during the period of its intrauterine formation, as well as with complications during the birth process.
The development of this pathology is unpredictable, and the consequences are also different.
A minimal loss of brain functionality in children can lead to the child’s inattention, with the inability to concentrate and remember the necessary information - this is a mild degree of consequences of the hypoxic-ischemic type of pathology. More severe complications lead to complete paralysis of the child’s body.
The hypoxic-ischemic type of foci of leukopathy in the brain also includes the perinatal form of leukoencephalopathy.
This encephalopathy develops according to the principle of an adult disease, only there is the only difference - its occurrence occurred in the womb, or in the first time immediately after birth.
Hemorrhagic type of leukoencephalopathy
This type of brain pathology occurs from vitamin deficiency of brain cells. Lack of vitamin thiamine leads to the development of multifocal leukoencephalopathy of a hemorrhagic nature.
This type of pathology proceeds in the same way as the development of other types of encephalopathy, but the etiology of this type is:
- Pathology in the digestive system that provoked anorexia;
- Prolonged gag reflex and a large amount of vomit from the body;
- Hemodialysis;
- Acquired immunodeficiency syndrome (AIDS).
Progressive multifocal leukoencephalopathy
This pathology is a deadly type of disease and is caused by papillomavirus. Quite often leads to death. This is a pathology that develops in more than 50.0% of AIDS patients.
A progressive multifocal form of leukoencephalopathy manifests itself:
- Body paralysis;
- Hemianopia of unilateral type;
- Peripheral paresis;
- Defect of personality consciousness;
- Syndromes of expiramidal types.
Disability with this type of pathology comes quite quickly, since its development occurs rapidly against the background of reduced immunity. There is a decrease in the functionality of the motor system, speech and hearing systems.
As the disease progresses, paralysis of parts of the body and partial paralysis of the brain occurs.
Brain damage in progressive multifocal leukoencephalopathy
Periventricular form
This type of pathology occurs from brain hypoxia with chronic insufficiency of blood in the cerebral vessels. Areas of ischemic damage are located not only in the white medulla, but also in the cells of the gray matter.
Localization of this destruction occurs in:
- Cerebellum;
- Bilateral pathology in the frontal regions of the cerebral cortex;
- In the brain stem.
All parts of the brain that are affected affect the development of motor functions. Disorder of these areas leads to paralysis of some parts of the body.
In newborn children, this type of leukoencephalopathy develops a pathology - cerebral palsy. This happens a few hours after the baby is born.
Leukoencephalopathy, in which white medulla disappears
This type is diagnosed in childhood from 2 calendar years to the 6th birthday. This disappearance occurs in the cerebral cortex due to gene mutation. This pathology has a single nonspecific focus, or small-focus lesions that affect all parts of the brain.
Symptoms of this type:
- No coordination in movement;
- Paresis of limbs;
- Decreased memory or loss of memory;
- Visual impairment - the nerve of the optic organ atrophies;
- Epilepsy attacks.
Such children have problems with food consumption, they are highly excitable, and also have increased muscle tone.
The pathology manifests itself in apnea, muscle cramps and a coma, which often ends in death.
How long do people live with leukoencephalopathy?
This pathology is the most dangerous disease of brain cells. With a stable course of the pathology, life time is measured according to medical forecasts to be a little more than two calendar years.
In the acute course of the disease, which immediately turned into a complicated form - no more than 30 calendar days.
The average life expectancy for a diagnosis of leukoencephalopathy is no more than 6 calendar months from the moment the exact type of pathology diagnosis is established. In this disease, time can decide the outcome of life - in a positive direction, or lead to death.
The faster the diagnosis is made and the cause of the disease is found, the sooner therapy can begin and save a person’s life.
Forecast
Multifocal progressive leukoencephalopathy is characterized by a steady deterioration of the clinical picture - neurological symptoms become more severe, up to the appearance of coma.
Currently, the disease is recognized by experts as incurable . In the acute form, life expectancy is no more than 10-12 months from the moment of diagnosis.
The prognosis can be considered relatively favorable with timely recognition of the pathology, taking adequate therapeutic measures - boosting immunity, powerful antiviral therapy.
The forecast is influenced by factors:
- age,
- the initial state of his health,
- accompanying illnesses,
- availability of specialized medical care,
- qualification of the doctor involved in treatment,
- the effectiveness of the therapy and the response of the human immune system to it.
In a number of cases, doctors were able to prolong a person’s life to 2.5-3 years, but the disease also had a lightning-fast course - death by the end of the month from the onset of leukoencephalopathy.
Features of the disease
Leukoencephalopathy is an incurable disease of the brain that affects the white matter of the brain. This pathology is a focal lesion, as well as a multifocal lesion of the white matter in the brain.
The etiology of the disease is viruses that are destructive to the body and primarily affect brain cells.
The occurrence of pathology occurs from reduced functionality of the immune system, mainly in very old people, as well as when the body is affected by the pathology of immunodeficiency. With AIDS, leukoencephalopathy develops in any age category.
There is a problem in drug treatment for this type of brain disease.
The thing is that there is a barrier in the brain through which only drugs containing fats can enter the brain cells.
These fat-soluble drugs can affect brain cells, but drugs that can effectively and quickly cure leukoencephalopathy are water-based. Water-soluble medications are not able to cross the brain barrier.
Therefore, to date, pharmacological companies have not been able to develop drugs for medically effective treatment of pathology - leukoencephalopathy.
Treatment tactics
Due to the difficulties of timely recognition of multifocal progressive leukoencephalopathy, the peculiarities of the mechanism of its appearance and course, specific treatment has not currently been developed.
Experts have made attempts to suppress viral activity with different subgroups of medications - interferons, immunostimulants. In some cases, a positive result was achieved after taking Cytarabine. However, numerous attempts to achieve a stable, long-term result were unsuccessful.
However, doctors are looking for new methods to combat viral pathology - for example, with the help of the antidepressant Mirtazapine. The drug blocks the spread of viral particles into neuroglial cells.
Attempts have been made to use:
- antiviral drugs,
- antitumor drugs,
- serotonin receptor antagonists.
There were no significant improvements in the immune system. Attempts to transplant bone marrow stem cells did not produce the desired results.
A separate group of specialists recognizes an effective method of restoring cellular immunity in people with an autoimmune variant of the disease by reducing the doses of immunosuppressants. Sometimes it is possible to completely stop therapy with such medications.
An exception is the condition after organ transplantation. As confirmation, doctors cite cases of pronounced regression of negative symptoms, and even recovery of patients after discontinuation of drugs from the subgroup of cytostatics.
Signs of development of leukoencephalopathy
Signs of many types of leukoencephalopathy appear gradually. At the beginning of development, attacks of forgetfulness and absent-mindedness appear. A person has difficulty remembering information and pronouncing long and complex words.
There is a constant feeling of self-pity, and the patient cries a lot. The intellectual performance of the brain is significantly reduced.
With the further development of the pathology, insomnia appears, which can alternate with an endless desire to sleep. Muscle tone increases, which together leads to groundless irritability of the patient.
At this stage of the development of the disease, severe tinnitus appears, as well as involuntary twitching of the optic nerve, which leads to unreasonable movement of the pupils.
If you do not start complex therapy at least at this stage, then the disease will lead to:
- Pathologies of psychoneurosis;
- To muscle fiber spasms;
- To dementia;
- To partial memory loss;
- To the pathology of dementia.
Symptoms of leukoencephalopathy
Symptoms of this pathology develop suddenly and progress at a rapid pace, which can lead the patient to the following signs of the disease:
- Bulbar type palsy;
- Parkinson's syndrome;
- Disturbed gait;
- Hand tremors occur;
- Signs of body trembling appear.
Patients with such symptoms are not aware of their pathology and brain damage, so it is necessary for relatives to promptly force such people to undergo a diagnosis in order to know how to treat the disease.
Diagnostics
To establish a diagnosis of leukoencephalopathy of brain tissue cells, it is necessary to undergo a number of diagnostic tests:
- Visual examination by a neurologist and medical history;
- Laboratory clinical blood test (general);
- Analysis of blood composition for the presence of psychotropic elements, alcohol, and drug-containing substances in it;
- MRI and CT (computed tomography, or magnetic resonance imaging) to identify lesions in parts of the brain;
- Instrumental diagnostics using electroencephalography will reveal a decrease in the brain activity of organ cells;
- Dopplerography is a technique that detects pathology and disorders in the blood flow system, as well as cerebral vessels;
- PCR analysis to detect the virus in the body. This analysis determines the DNA of the provoking virus;
- Brain cell biopsy;
- Cerebrospinal fluid puncture.
If it is revealed that viruses are the provocateur of leukoencephalopathy, then further diagnosis is carried out using electron microscopy of brain cells.
Dopplerography of the vessels of the head
Differential diagnostic examination is carried out for the following pathologies:
- The disease is toxoplasmosis;
- Pathology cryptococcosis;
- Dementia due to HIV;
- Leukodystrophy disease;
- CNS lymphoma disease;
- Pathology subacute sclerosing panencephalitis;
- Multiple sclerosis.