Миопатия является врожденной патологией, вызванной генной мутацией. Механизм развития заболевания до конца не изучен, поэтому предсказать вероятность рождения больного ребенка не представляется возможным. Случается, что у двух абсолютно здоровых людей рождается ребенок с какой-либо формой миопатии. Установлено, что причиной является нарушение обмена в мышечных тканях, вследствие чего они теряют креатин и истощаются.
Миопатия Беккера, или доброкачественная псевдогипертрофическая миопатия, – наиболее легкая форма заболевания, которая впервые была описана в 1955 году Беккером и Кинером. Установлена генетическая общность миопатии Беккера и Дюшенна – обе разновидности вызывают аллельные мутации одних и тех же генов. Характерной особенностью дистрофии Беккера является то, что болеют только лица мужского пола, на 20 тыс. новорожденных приходится 1 заболевший ребенок.
Мышечная дистрофия Дюшенна-Беккера
OMIM 310200
Наша команда профессионалов ответит на ваши вопросы
Миодистрофия Дюшенна (МДД) — наследственное заболевание, которое начинается в возрасте 2-5 лет и характеризуется прогрессирующеймышечной слабостью, атрофией и псевдогипертрофией проксимальных мышц, нередко сопровождается кардиомиопатиями и нарушением интеллекта. На ранних этапах заболевания наблюдается повышенная утомляемость при ходьбе, изменение походки («утиная походка»). При этом происходит постепенная деградация мышечных тканей. 95% больных перестают ходить в возрасте 8-12 лет. В возрасте 18-20 лет больные, как правило, умирают, часто от дыхательной недостаточности. Выделяют аллельную МДД форму – мышечную дистрофию Беккера (МДБ, OMIM 310200), которая характеризуется сходными клиническими проявлениями, более поздним началом (примерно в 10-16 лет) и более мягким течением. Такие больные часто сохраняют способность ходить до 20 лет, а некоторые – до 50-60 лет, хотя в патологический процесс вовлечены те же мышцы, что и при МДД. Продолжительность жизни таких больных сокращена незначительно.
Биохимическим маркером заболевания является повышенный (в 100-200) раз уровень креатинфосфокиназы (КФК) в крови. У носительниц поврежденного гена уровень КФК в среднем также несколько повышен.
Тип наследования мышечной дистрофии Дюшенна – Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена DMD, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Мышечная дистрофия Дюшена (МДД) встречается приблизительно у одного из 2500-4000 новорожденных мальчиков.
Ген DMD, ответственный за прогрессирующую мышечную дистрофию Дюшена/Беккера (МДД/МДБ), находится в локусе Хр21.2, имеет размер 2,6 млн п.н. и состоит из 79 экзонов. В 60% случаев мутации, приводящие к МДД/МДБ, представляют собой протяженные делеции (от одного до до нескольких десятков экзонов), в 30% случаев – точковые мутации и в 10% случаев – дупликации. Из-за наличия так называемых «горячих участков» делеций амплификация 27 экзонов и промоторной области гена DMD позволяет выявлять примерно 98% всех крупных делеций. Поиск точковых мутаций затруднен из-за большого размера гена и отсутствия мажорных мутаций.
В Центре Молекулярной Генетики проводится измерение уровня КФК в крови, а также прямая диагностика МДД/МДБ, представляющая собой поиск крупных делеций/lдупликаций во всех экзонах гена DMD и поиск «точковых» мутаций гена DMD методом NGS (next generation sequensing). Исследование методом NGS позволяет так же выявлять делеции всех экзонов гена DMD у больных мальчиков. Анализ всех экзонов гена позволяет определить точные экзонные границы делеции в сучае ее выявления, и таким образом, установить приводит ли данная делеция к сдвигу рамки считывания белка, что в свою очередь важно для прогноза формы заболевания — миодистрофия Дюшенна или Беккера. Таким образом, сочетание различных методов исследования позволяет выявлять практически все мутации гена DMD.
Наличие любого типа мутаций (делеции/дупликации в одном или нескольких экзонах, «точковые» мутации) является молекулярно-генетическим подтверждением клинического диагноза миодистрофии Дюшена/Беккера и позволяет проводить дородовую диагностику в данной семье.
Внимание! Для измерения уровня КФК кровь должна быть свежей (не замороженной)!
В случае дородовой диагностики необходим биоматериал плода, в качестве которого можно использовать ворсины хориона (с 8-й до 12-й недели беременности), амниотическую жидкость (с 16-й до 24-й недели беременности) или пуповинную кровь (с 22-й недели беременности).
Нами разработаны наборы для ДНК-диагностики прогрессирующей мышечной дистрофии Дюшенна / Беккера. Наборы предназначены для использования в диагностических лабораториях молекулярно-генетического профиля.
Код по МКБ-10
Под миокардиодистрофией понимают специфический недуг сердца, имеющий не воспалительный характер. Он характеризуется расстройствами обменных процессов, которые протекают в миоцитах. При этом изменяются сократительные способности сердечной мышцы, появляется сердечная недостаточность.
Классификация заболеваний МКБ-10 — это нормативный документ, который содержит коды для заболеваний с систематизацией по разделам. Это международная разработка, которая облегчает сбор и хранение информации для создания статистики по всем странам. МКБ-10 создана Всемирной организацией здравоохранения. Документ пересматривается каждые 10 лет, в него всегда вносятся дополнения. Если раньше для миокардиодистрофии был отдельный код, то сейчас он не присваивается этому заболеванию по МКБ-10. Однако врачи могут поставить номер I.42, который подразумевает кардиомиопатию. Несмотря на схожесть названий, кардиомиопатия и миокардиодистрофия считаются разными явлениями, так как первое предполагает более широкий спектр различных сердечных патологий, которые не связаны с болезнями сердца и сосудистой системой. Поэтому эти термины нельзя использовать в качестве синонимов. Но для миокардиодистрофии все же установлен один код по МКБ-10, который предполагает только кардиомиопатию с метаболическими нарушениями и проблемами питания.
В целом, раздел I42 предполагает кардиомиопатию, но исключается состояние, которое осложнено беременностью и послеродовой ишемической болезнью. Номер I42.0 предполагает дилатационную разновидность, а 42.1 — это гипертрофичекая обструктивная патология. Под номером 42.2 подразумевают другие формы гипертрофической патологии. Код 42.3 — это эндомиокардиальное заболевание, а 42.4 — фиброэластоз эндокардиального типа. Если у пациента обнаруживаются другие рестриктивные сердечные патологии, то устанавливается номер 42.5. Если кардиомиопатия имеет алкогольное происхождение, то предполагается код 42.6. Если заболевание вызвано влиянием лекарственных препаратов или другими внешними факторами, то устанавливается номер 42.7. Прочие кардиомиопатии имеют номер 42.8, а если формы патологии не уточнены, то код 42.9.
Разница между мышечной дистрофией Дюшенна и Беккера
Ключевое отличие мышечной дистрофии Дюшенна и мышечной дистрофии Беккера, заключается в том, что при мышечной дистрофии Дюшенна дистрофин отсутствует, тогда как при мышечной дистрофии Беккера дистрофин присутствует, хотя и на низких уровнях. В этом ключевое отличие мышечной дистрофии Дюшенна и Беккера. Другим важным отличием между этими двумя условиями является уровень их серьезности.
Мышечная дистрофия Дюшенна и мышечная дистрофия Беккера являются X-сцепленными рецессивными расстройствами, характеризующимися прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией мышечных волокон и характеризующимися изменениями уровней дистрофина. Мышечные дистрофии — это наследственные прогрессирующие мышечные нарушения, возникающие в результате дефектов одного или нескольких генов, необходимых для нормальной структуры и функции мышц. Дистрофия Дюшенна и дистрофия Беккера являются второй по распространенности мышечной дистрофией (после фасцикапулохимеральной мышечной дистрофии). Они вызваны мутациями гена дистрофина, самого большого известного гена человека, в локусе Xp21.2. При дистрофии Дюшенна эта мутация приводит к серьезному отсутствию ( Обзор и основные отличия
- Что такое мышечная дистрофия Дюшенна
- Что такое мышечная дистрофия Беккера
- Сходство между мышечной дистрофией Дюшенна и Беккера
- В чем разница между мышечной дистрофией Дюшенна и Беккера
- Заключение
Что такое мышечная дистрофия Дюшенна?
Мышечная дистрофия Дюшенна или миодистрофия Дюшенна (МДД) — представляет собой Х-сцепленное рецессивное заболевание, характеризующееся прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией мышечных волокон а также отсутствием структурного стержневидного белка — дистрофина, который необходим для стабильности клеточной мембраны. Это заболевание вызвано мутациями гена дистрофина, самого большого известного гена человека, в локусе Xp21.2. При дистрофии Дюшенна эта мутация приводит к сдвигу рамки считывания и к серьезному отсутствию ( Это заболевание поражает одного из трех тысяч младенцев мужского пола.
Мышечная дистрофия Дюшенна
Это расстройство обычно проявляется в возрасте от 2 до 3 лет. Слабость поражает проксимальные мышцы, обычно на нижних конечностях. Дети часто ходят пешком, имеют походку и лордоз. Им трудно бегать, прыгать, подниматься по лестнице и даже с пола. Дети часто падают, часто вызывая переломы рук или ног (примерно у 20% пациентов). Прогрессирование слабости является устойчивым, и контрактуры сгибания конечностей и сколиоз развиваются почти у всех детей. Развивается твердая псевдогипертрофия (жировая и фиброзная замена определенных расширенных групп мышц). Большинству детей необходимо использовать инвалидную коляску уже к 12 годам и в итоге они умирают от респираторных осложнений в возрасте 20 лет.
Последствия поражения сердечной мышцы включают дилатационную кардиомиопатию , нарушения проводимости и аритмии. Такие осложнения возникают примерно у трети пациентов в возрасте 14 лет и у всех пациентов старше 18 лет. однако, поскольку эти пациенты не могут заниматься спортом, поражение сердца обычно протекает бессимптомно до поздней стадии заболевания. Около трети больных имеют слабые, непрогрессирующие умственные нарушения, которые влияют на речевые способности больше, чем на производительность.
Мутационный анализ ДНК из лейкоцитов периферической крови является основным подтверждающим тестом. Он может выявить отклонения в гене дистрофина (делеции примерно у 70% пациентов с дистрофией Дюшенна и у 85% пациентов с дистрофией Беккера; дупликации примерно у 10% обеих групп).
Если генетическое тестирование не подтверждает диагноз, следует провести анализ дистрофина с иммуноокрашиванием образцов биопсии мышц. Дистрофин не обнаруживается у пациентов с дистрофией Дюшенна.
Пациенты с дистрофией Дюшенна должны иметь базовую оценку сердечной функции с помощью ЭКГ и эхокардиографии на момент постановки диагноза или в возрасте 6 лет.
Выявление носителей и пренатальная диагностика возможны при использовании традиционных исследований (например, анализ родословной, определение CK, определение пола плода) в сочетании с анализом рекомбинантной ДНК и иммуноокрашивания дистрофина мышечной ткани.
Лечение
Дистрофия Беккера является неизлечимым заболеванием, и пациенты получают преимущественно симптоматическую и метаболическую терапию. Однако ученые ведут активные поиски эффективных методов лечения, включая генные и клеточные. В настоящее время можно рассчитывать только на поддержание двигательной способности и самостоятельности пациента. Лечебные препараты, способные замедлить прогрессирование мышечной дистрофии:
- актопротекторы (Этилтеобензимидазол). Стимулируют физическую работоспособность и препятствуют утомлению;
- ингибиторы холинэстеразы (Неостигмин). Способствуют нервно-мышечной проводимости, повышают тонус гладких мышц;
- аденозинтрифосфорная кислота, или сокращенно АТФ. Применяется для улучшения трофики мышц внутримышечным введением 1-2 раза в день. Курс лечения – 30-40 инъекций, после месячного перерыва курс при необходимости повторяют;
- анаболические стероиды (Метиладростендиол). Использование лекарств этой группы помогает ускорить развитие и обновление клеток, тканей и мышц;
- сердечные препараты;
- витамины группы В и Е.
Медикаментозную терапию миопатии сочетают с физиотерапией, массажем и ЛФК. Комплекс мероприятий составляется с учетом посильной физической нагрузки на пациента, чтобы избежать чрезмерного перенапряжения ослабленной мускулатуры. В определенных случаях больные нуждаются в консультации ортопеда, который поможет подобрать специальные корректирующие устройства – корсеты, обувь и пр.
Мануальная терапия – один из способов коррекции состоянии пациента
Хирургическое вмешательство проводится в случаях, когда пациент испытывает слишком болезненные ощущения в сухожилиях мышц – операция способствует их удлинению путем коррекции контрактур.
Симптомы миодистрофии
- Проблемы с мышечной функцией
: Часто кто-либо из членов семьи замечает, что с ребенком что-то не в порядке. Мальчики, заболевшие
мышечной дисторфии Дюшена
, начинают ходить позже сверстников. У них увеличены икроножные мышцы; они испытывают затруднения при беге, прыжках, подъеме по лестнице. Они часто падают, у них может появиться манера ходить на носках. Кроме того, у них бывает задержка речи. Один из классических симптомов
мышечной дисторфии Дюшена
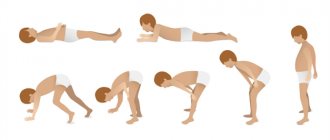
– это так называемая положительная проба Говерса, которая проявляется в том, что для перехода из положения лежа в положение стоя, мальчик вынужден перебирать кистями рук и локтями снизу вверх по телу. Он вынужден делать это из-за слабости тазобедренных мышц. - ВЫСОКОЕ СОДЕРЖАНИЕ КРЕАТИНКИНАЗЫ
(КК) В МЫШЕЧНОМ БЕЛКЕ, ВЫЯВЛЕННОЕ ПРИ АНАЛИЗЕ КРОВИ. Обнаружение высокого содержания креатинкиназы должно послужить сигналом и срочно обратиться к специалисту для подтверждения диагноза. Высокий показатель КК наблюдается у людей, страдающих и другими видами мышечных заболеваний, а потому для подтверждения
мышечной дисторфии Дюшена
одного только показателя креатинкиназу недостаточно. - ВЫСОКОЕ СОДЕРЖАНИЕ ТАК НАЗЫВАЕМЫХ «ПЕЧЕНОЧНЫХ» ФЕРМЕНТОВ
(AST И ALT), ВЫЯВЛЕННОЕ ПРИ АНАЛИЗЕ КРОВИ. Их высокое содержание в крови нередко связывают с заболеваниями печени, однако оно может быть вызвано и миодистрофией. При неожиданном выявлении высокой концентрации этих ферментов без какой-либо иной причины следует насторожиться. Уровень реатинкиназы может быть также высоким, а потому подозрение на диагноз «миодистрофия» не лишено оснований. - ЗАДЕРЖКА РЕЧЕВОГО РАЗВИТИЯ. У детей, страдающих мышечной дисторфии Дюшена
, нередко отмечается некоторое отставание в развитии речи, на что также можно обратить внимание
Классификация
Алиментарная дистрофия имеет код Е41 по МКБ 10.
В гастроэнтерологии заболевание принято разделять по тяжести и по формам. По форме выделяют сухую (кахектическую) и отечную дистрофию. В кахектической форме недуг имеет более опасное течение. Отечная же форма характеризуется распространенными отеками, включая внутренние (плеврит, перикардит и асцит), данная форма легче поддается лечению.
Также различают 3 стадии тяжести заболевания:
- Первая стадия
характеризуется незначительным снижением веса и сохранением работоспособности с жалобами на повышение аппетита, жажду и частые мочеиспускания, слабость и зябкость. - Вторая стадия
характеризуется существенным исхуданием со снижением работоспособности. Пациенты с заболеванием на этой стадии все еще в состоянии себя обслуживать, однако практически не могут трудиться. Возможны отеки, отмечается значительное снижение уровня белка, зачастую понижается уровень глюкозы в крови. - Третья стадия
характеризуется резким истощением и неспособностью больного самостоятельно передвигаться.
Генетический анализ.
Проведение генетического теста
необходимо в любом случае, даже если диагноз
мышечной дисторфии Дюшена
подтвержден результатами биопсии мышц. С помощью различных видов генетического анализа можно получить конкретные и более подробные сведения об изменениях (мутациях) в ДНК. Располагать генетическим подтверждением диагноза важно по нескольким причинам: можно определить может ли мальчик участвовать в ряде клинических исследований, касающихся определенной мутации, кроме того, это помогает семье принять решения, связанные с пренатальной диагностикой и будущей беременностью. на данный момент известно о более 900 видах мутаций, поэтому так важна точная диагностика дистрофии в Израиле, даже если вы делали этот тест на Родине.
Как только стала известна точная мутация или изменение в ДНК гена дистрофина матерям
нужно дать возможность пройти генетический анализ с целью выяснить, являются ли они носителями дефектного гена или нет. Эта информация будет важна для родственниц по материнской лини (сестры, дочери, тети, кузины), поскольку позволит выяснить, не являются ли они сами носителями дефектного гена. Генетическая диагностика, разъясненная
генетиком
-консультантом поможет семье лучше понять результат теста и его потенциальное воздействие, которое он может окажет на всех членов семьи.
Диагностика
Диагноз может быть поставлен на основании осмотра пациента и сбора анамнеза. Если анамнез содержит указания на продолжительное голодание, имеется характерная симптоматика и показатели лабораторных анализов (развернутый биохимический и клинический анализы крови, наблюдаются признаки дистрофии органов по МРТ, УЗИ, КТ), а также исключены другие заболевания, то ставится диагноз алиментарная дистрофия.
Дифференцировать заболевание следует с прочими патологиями, которые приводят к истощению: онкологические процессы (рак кишечника, желудка и прочие патологические состояния), сахарный диабет, туберкулез, тиреотоксикоз и гипофизарные расстройства.
Алиментарная дистрофия отличается от других болезней выраженным усилением аппетита и жажды, голодом, изменениями кожи и сильным истощением мышц, брадикардией и понижением температуры тела, а также нарушением работы эндокринных желез.
Биопсия мышцы.
Ваш врач может порекомендовать сделать биопсию мышцы (т.е. взять на анализ маленький образец мышцы). Наличие генетической мутации при мышечной дисторфии Дюшена
означает, что организм либо не может вырабатывать белок дистрофина, либо вырабатывает его, но в недостаточном количестве. С помощью анализа биопсии мышцы можно выяснить какое количество дистрофина, находится в мышечных клетках.
Если подтверждение диагноза уже получено при генетическом анализе, биопсия мышцы необязательна
.
Биопсия не отменяет генетический тест
!
При исследовании данных биопсии мышцы обычно проводятся два вида анализов: иммуно-гистохимический
и
иммуноблоттинг-анализ
(метод исследования белковых антигенов) на дистрофин. Эти тесты позволяют определить наличие или отсутствие дистрофина в количественной форме; с их помощью можно отличить
мышечной дисторфии Дюшена
от более мягкой формы миодистрофии МДБ.
Причины
Причиной алиментарной дистрофии служит продолжительное голодание, когда в организм человека поступает недостаточное количество необходимых питательных веществ, причем учитывается относительная их недостаточность: если поступление калорий отличается от их расхода.
Голодание возможно по различным причинам (экологическое бедствие война, и прочие случаи вынужденного длительного недостатка пищи; диеты; рубцы и сужение пищевода и др.), однако процесс усугубляется переохлаждением и тяжелым физическим трудом.
Следует отметить, что развитие алиментарной дистрофии возможно лишь в случае продолжительного энергетического голодания. В организме при этом сначала истощаются запасы жиров и гликогена, затем на обеспечение процессов обмена идут запасы внутритканевого белка. Процессы дистрофии в первую очередь протекают в коже и мышцах, после этого заболевание распространяется на внутренние органы, и в последнюю очередь – на жизненно важные органы (мозг, почки и сердце).
На последних стадиях истощаются запасы минералов и витаминов, перестает функционировать иммунитет. Летальный исход наступает обычно в результате сердечной недостаточности либо присоединившейся инфекции после угнетения иммунной системы.