Нервно-мышечные заболевания: этимология, классификация, симптомы и лечение
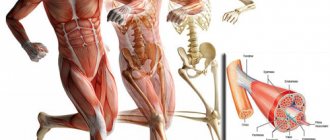
Нервно-мышечные заболевания (НМЗ) – одна из самых многочисленных групп наследственных заболеваний, характеризующаяся нарушением функций произвольной мускулатуры, снижением или утратой контроля за движениями. Возникновение этих заболеваний обусловлено дефектом эмбрионального развития или генетически детерминированной патологией.
Характерным проявлением наследственных нервно-мышечных заболеваний является атаксия – расстройство координации движений, нарушение моторики. При статической атаксии нарушается равновесие в состоянии стоя, при динамической – координация во время движения.
Для нервно-мышечных заболеваний характерны следующие симптомы: слабость, атрофия мышц, самопроизвольные мышечные подёргивания, спазмы, онемение и т.п.
При нарушении нервно-мышечных соединений у больных может наблюдаться опущение век, двоение в глазах, и ещё ряд проявлений ослабевания мышц, которые в течение дня только усиливаются.
В отдельных случаях возможны нарушение глотательной функции и дыхания.
Классификация нервно-мышечных заболеваний
Нервно-мышечные заболевания можно отнести к четырем основным группам в зависимости от места локации:
- мышц;
- нервно-мышечных окончаний;
- периферических нервов;
- двигательного нейрона.
По виду и типу нарушений их разделяют на следующие основные группы:
- первичные прогрессирующие мышечные дистрофии (миопатии);
- вторичные прогрессирующие мышечные дистрофии;
- врожденные не прогрессирующие миопатии;
- миотонии;
- наследственные пароксизмальные миоплегии.
Миопатия
Термин миопатия (миодистрофия) объединяет достаточно большую группу заболеваний, которые объединены общим признаком: первичным поражением мышечной ткани. Развитие миопатии могут спровоцировать различные факторы: наследственность, вирусное поражение, нарушение обмена веществ и ряд других.
К воспалительным миопатиям (миозитам) относятся заболевания, вызванные воспалительным процессом. Они развиваются в результате аутоиммунных нарушений и могут сопровождаться другими заболеваниями аналогичной природы. Это дерамтомизит, полимиозит, миозит с различными включениями.
Митохондриальные миопатии. Причиной возникновения заболевания являются структурные или биохимические митохондрии. К этому типу заболеваний относятся:
- синдром Кернса-Сайре;
- митохондриальная энцефаломиопатия;
- миоклонус эпилепсия с «разорванными красными волокнами».
Помимо этих заболеваний, существует ещё ряд редко встречающихся видов миопатий, поражающих центральный стержень, эндокринную систему и т.д.
При активном течении миопатия может привести к инвалидности и дальнейшему обездвиживанию больного.
Вторичные прогрессирующие мышечные дистрофии
Заболевание связано с нарушением работы периферических нервов, нарушением снабжения органов и тканей нервными клетками. В результате происходит дистрофия мышц.
Различают три вида вторичной прогрессирующей мышечной дистрофии: врождённую, раннюю детскую и позднюю. В каждом случае болезнь протекает с большей или меньшей степенью агрессии. Для людей с таким диагнозом средняя продолжительность жизни составляет от 9 до 30 лет.
Врождённые не прогрессирующие миопатии
К ним относятся наследственные не прогрессирующие или слабо прогрессирующие заболевания мышц, диагностированные во внутриутробный период или сразу поле рождения младенца. Основной признак – гипотония мышц с явно выраженной слабостью. Это заболевание иначе называют «синдром вялого ребёнка», что точно характеризует его состояние.
В большинстве случаев поражается область нижних конечностей, реже – верхних, в исключительных случаях встречается поражение краниальной мускулатуры – нарушение мимики, движения глаз.
В процессе развития и роста ребёнка отмечаются проблемы с моторикой, дети часто падают, поздно начинают сидеть и ходить, не могут бегать и прыгать. При этом нет нарушений интеллекта. К сожалению, этот тип миопатий неизлечим.
Симптомы
При всех видах миопатий основным симптомом является слабость мышц. Чаще всего поражениям подвержены мышцы плечевого пояса, бедер, области таза, плечи.
Для каждого типа характерно поражение конкретной группы мышц, что важно учитывать при диагностике.
Поражение происходит симметрично, поэтому он способен производить действия поэтапно, постепенно включая в работу различные участки.
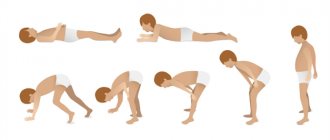
При поражении ног и области таза, для того, чтобы встать с пола нужно сначала опереться руками о пол, встать на колени, взяться за опору и после этого больной может сесть на стул или кровать. Самостоятельно, не прибегая к помощи рук, он встать не сможет.
При миопатии реже всего встречаются случаи поражения мышц лица. Это птоз (опущение верхнего века), опускается верхняя губа. Возникают проблемы с речью, вызванные нарушением артикуляции, возможно нарушение глотательной функции.
Большинство миопатий протекают с практически одинаковыми признаками. С течением времени происходит атрофия мышечной ткани, на фоне которой активно разрастается соединительная.
Визуально это выглядит как натренированные мышцы – т.н. псевдогипертрофия. В самих суставах происходит формирование контрактуры, стягивается мышечно-сухожильное волокно.
В результате появляются болевые ощущения и происходит ограничение подвижности сустава.
Миоплегия
Как и миопатии, это наследственные нервно-мышечные заболевания, для которых характерны приступы мышечной слабости или паралича конечностей. Различают следующие разновидности миоплегий:
- гипокалиемическую;
- гиперкалиемическую;
- нормокалиемическую.
Приступ миоплегии вызывается перераспределением калия в организме – происходит его резкое уменьшение в межклеточной жидкости и плазме, и увеличение (переизбыток) в клетках.
В мышечных клетках происходит нарушение поляризации мембран, происходит изменение электролитических свойств мышц.
Во время приступа у больного возникает резкая слабость конечностей или туловища, возможны проявления в глотке, гортани, воздействие на дыхательные пути. Это может привести к летальному исходу.
Миастения
Заболеванию чаще всего подвержены женщины (2/3 от общего количества больных). Миастения имеет две формы – врождённую и приобретённую. При этом заболевании происходит нарушение передачи нервных импульсов, в результате чего возникает слабость в поперечнополосатых мышцах.
Заболевание связано с изменением функций нервно-мышечной системы. Слабеющие мышцы воздействуют на нормальную работу органов:у больного могут быть постоянно полузакрыты веки, происходит нарушение мочеиспускания, возникают трудности при жевании, ходьбе. В результате болезнь может привести к инвалидности и даже смерти.
Болезни двигательного нейрона (БДН)
Для болезней двигательного нейрона характерно поражение двигательных нейронов головного и спинного мозга. Постепенное отмирание клеток влияет на функцию мышц: они постепенно ослабевают, а зона поражения увеличивается.
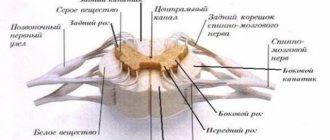
Нейроны головного мозга, отвечающие за движение, расположены в коре головного мозга. Их ответвления – аксоны – спускаются в область спинного мозга, где и происходит контакт с нейронами этого отдела. Этот процесс называют синапсом.
В результате нейрон головного мозга выделяет особое химическое вещество (медиатор), передающий сигнал нейронам спинного мозга.
Эти сигналы отвечают за сокращение мышц различных отделов: шейного, грудного, бульбарного, поясничного отделов.
В зависимости от выраженности повреждения нейронов и их локализации выделяют несколько видов БНД. Во многом проявления заболеваний одинаковы, но по мере прогрессирования болезни разница становится всё существеннее.
Выделяют несколько различных видов БНД:
Боковой амиотрофический склероз
Это один из четырёх основных видов болезни двигательного нейрона. Она встречается у 85% пациентов, у которых диагностировано заболевание двигательного нейрона. Областью поражения могут быть как нейроны головного мозга, так и спинного. В результате происходит атрофия мышц и их спастичность.
При БАС отмечается слабость и нарастающая усталость в конечностях. У некоторых людей отмечается слабость в ногах во время ходьбы и слабость в руках, при которой невозможно удержать в руках вещи.
В большинстве случаев заболевание диагностируется в возрасте до 40 лет, при этом заболевание абсолютно не затрагивает интеллект. Прогноз для больного, которому поставлен диагноз БАС не самый благоприятный – от 2 до 5 лет. Но встречаются и исключения: наиболее известный из всех человек, который прожил с этим диагнозом более 50 лет – профессор Стивен Хокинг.
Прогрессирующий бульбарный паралич
Связан с нарушением речи и глотания. Прогноз с момента постановки диагноза составляет до трёх лет с момента постановки диагноза;
Первичный литеральный склероз
Затрагивает только нейроны головного мозга и поражает нижние конечности. В редких случаях сопровождается нарушениями движений рук или проблемами с речью. В более поздних стадиях может перейти в БАС.
Прогрессирующая мышечная атрофия
Возникает при поражении двигательных нейронов спинного мозга. Первые проявления выражаются в слабости рук. Прогноз по этому заболеванию составляет от 5 до 10 лет.
Диагностика
Для установления точного диагноза важно провести следующие исследования:
- биохимические. Определение мышечных ферментов, прежде всего креатинфосфокиназа (КФК). Определяется уровень миоглобина и альдолазы;
- электрофизиологические. Электромиография (ЭМГ) и электронейромиография (ЭНМГ) помогают в дифференцировании первичной и вторичной миопатией. Они также помогают выявить, что страдает первично – спинной мозг или периферический нерв;
- паталогоморфологические. Заключаются в проведении мышечной биопсии. Изучение материала также помогает дифференцировать первичную или вторичную миопатию. Определение уровня содержания дистрофина даёт возможность отличить миопатию Дюшена от миодистрофии Беккера, что важно для назначения правильного лечения;
- ДНК-диагностика.Исследование ДНК-лейкоцита позволяет выявить наследственные заболевания у 70 % больных.
Лечение нервно-мышечных заболеваний
При постановке одного из диагнозов, относящихся к нервно-мышечным заболеваниям, в каждом конкретном случае лечение подбирается индивидуально, с учётом всех полученных анализов. Пациент и его близкие изначально должны понимать, что это длительный и очень сложный процесс, требующий больших финансовых затрат.
Трудности назначения лечения связаны ещё и с тем, что не всегда возможно точно определить первичный дефект метаболизма. При этом заболевание постоянно прогрессирует, а значит, лечение в первую очередь должно быть направлено на замедление развития болезни. Это поможет сохранить возможность больного к самообслуживанию и повлиять на качество его жизни.
Методы лечения нервно-мышечных заболеваний
- Коррекция метаболизма скелетной мускулатуры. Назначаются препараты, стимулирующие метаболизм, препараты калия, комплексы витаминов, анболические стероиды;
- Стимуляция сигментарного аппарата. Нейростимуляция, миостимуляция, рефлексотерапия, бальнеотерапия, занятие лечебной физкультуры (упражнения и нагрузка подбираются индивидуально);
- Коррекция кровотока. Различные виды массажа, тепловые процедуры на определённые участки, оксигенобаротерапия;
- Соблюдение диеты и парентеральное питание для обеспечения организма всеми необходимыми питательными веществами – белком, солями калия, витаминами нужной группы;
- Корректирующие занятия у ортопеда. Коррекция контрактур, деформаций грудной клетки и позвоночника и т.п.
На сегодня не придумано лекарство, от приёма которого любой человек в один миг станет абсолютно здоровым. При всей сложности ситуации, для пациента с нервно-мышечным заболеванием важно продолжать максимально качественную жизнь. Пример Хокинга, который более 50 лет был прикован к инвалидному креслу, но продолжал проводить исследования, говорит о том, что болезнь не повод сдаваться.
Источник: //onevrologii.ru/nervno-myshechnye-patologii/nervno-myshechnye-zabolevaniya
Врождённые не прогрессирующие миопатии
К ним относятся наследственные не прогрессирующие или слабо прогрессирующие заболевания мышц, диагностированные во внутриутробный период или сразу поле рождения младенца. Основной признак – гипотония мышц с явно выраженной слабостью. Это заболевание иначе называют «синдром вялого ребёнка», что точно характеризует его состояние.
В большинстве случаев поражается область нижних конечностей, реже – верхних, в исключительных случаях встречается поражение краниальной мускулатуры – нарушение мимики, движения глаз.
Нервно-мышечные болезни – Мед Др
Нервно-мышечные заболевания являются наиболее многочисленной группой среди всех заболеваний нервной системы. К ним относятся различные виды миопатий, невральные и спинальные амиотрофии, миастения, миотония и периодический паралич.
Относительно высокая частота этих заболеваний, тяжелая инвалидизация при большинстве из них, поражение в детском или цветущем возрасте делают их весьма актуальными. Между тем практические врачи недостаточно полно знакомы с данной проблемой, особенно с достижениями в изучении патогенеза, современными диагностическими методами и новыми методами лечения.
Представляемая вниманию читателей монография создана сотрудниками двух коллективов, более четверти века изучающими различные аспекты патологии мышц; начало этому изучению было положено Н. И. Гращенковым, 3. Л. Лурье, Л. Б. Перельманом. Это нашло отражение в построении монографии и выборе материалов.
Исходя из относительно небольшого объема книги, авторы уделили основное внимание тем аспектам проблемы, в разработке которых они приняли непосредственное участие и имеют наибольший опыт.
В связи с тем, что книга является по существу коллективной монографией, в создании которой приняли участие авторы, принадлежащие к двум научным направлениям — клинико-генетическому, базирующемуся главным образом на использовании биохимических методов анализа, и клинико-патофизиологическому, применяющему в качестве инструмента анализа в основном электромиографические методы исследования, — при изложении отдельных проблем авторам не удалось полностью избежать противоречий в некоторых вопросах. Однако они и не стремились к однообразному изложению, оставляя возможность читателю создать собственное мнение после ознакомления с фактическим материалом.
Книга не является учебным пособием и справочным руководством по клинической миологии, поэтому напрасно искать в ней ответа на все интересующие читателей вопросы. Так, исходя из наличия имеющихся публикаций, авторы не рассматривают боковой амиотрофический склероз, спинальные амиотрофии, наследственные поражения периферической нервной системы; другие же разделы изложены более полно.
Наряду с обсуждением проблем прогрессирующих мышечных дистрофий описаны непрогрессирующие мышечные дистрофии, а также нервно-мышечные расстройства, возникающие при заболеваниях желез внутренней секреции и обмена веществ. Практика показывает, что нарушения двигательной функции задолго предшествуют другим симптомам эндокринных страданий.
Освещены вопросы клиники, диагностики и лечения воспалительных и аутоиммунных заболеваний нервно-мышечной системы. Ранняя диагностика этих заболеваний особенно важна, так как она обеспечивает проведение ранней терапии, способной в корне изменить течение и прогноз болезни.
В связи с этим лечение в данных разделах описано более подробно.
Изложению частных проблем предпосланы две главы, имеющие как теоретическое, так и чисто практическое значение: о роли биохимических методов исследования и клинической электромиографии в изучении механизмов развития и диагностике нервно-мышечных заболеваний.
Мы полагаем, что включение этих глав, написанных применительно к задачам клинической миологии, не только поможет читателю в понимании затронутых в монографии проблем, но и будет способствовать более широкому использованию данных методов в диагностике и контроле за течением патологических процессов.
Осуществление исследований, положенных в основу книги, потребовало участия большого числа сотрудников тех учреждений, где были выполнены эти исследования, — Института общей патологии и патологической физиологии АМН СССР, Клиники нервных болезней и факультетской хирургии I ММИ им И. М. Сеченова, Центральной клинической больницы № 3 МПС. Авторы приносят сердечную благодарность сотрудникам и руководителям этих учреждений, а также всем, кто советами и критическими замечаниями способствовал созданию книги.
«Нервно-мышечные болезни», Б.М.Гехт, Н.А.Ильина
Бадалян Л. О. Клинико-генетический анализ и вопросы классификации прогрессирующих мышечных дистрофий. — В кн.: Вопросы клинической нейрогенетики. М., 1973, с. 68 — 79. Бондаренко Е. С.
Наследственные мышечные дистрофии/ЦИУ. — М., 1976. Гаджиев С. А., Логель Л. В., Ваневский В. Л. Диагностика и хирургическое лечение миастении. — Л.: Медицина, 1971. Гаусманова-Петрусевич И.
Мышечные заболевания/Пер. с…
Adams R. Diseases of muscle: A stucfy in pathology. 3 ed. — 1975. Bradley W. G. Disorders of peripheral nerves. — Oxford; London, 1974. Buchthal F. An introduction to electromyography. — Copenhagen, 1957. Dubowitz V., Brooke M. H. Muscle biopsy: A modern approach. — London, Philadelphia, Toronto, 1973. Emeryk В., Strugalska M. Evaluation of results…
Миастенический синдром при терминальной полиневропатии
Особой формой миастенических расстройств, обусловленных поражением терминальных ветвлений двигательных нервов и грубыми расстройствами нервно-мышечной передачи, является описанная нами в 1979 г.
терминальная полиневропатия с миастеническим синдромом. К настоящему времени мы наблюдаем в течение 12 лет 13 больных с этим симптомокомплексом (12 мужчин и 1 женщина).
У 2 больных заболевание началось в возрасте старше 30 лет,…
Прежде всего при данном заболевании наблюдаются типичные ЭМГ-ские изменения, свидетельствующие о грубых изменениях нервно-мышечной передачи, снижение амплитуды вызванного ПД мышцы, грубый блок нервно-мышечной передачи при стимуляции редкими частотами (1 и 3 имп/с) и тетанизации мышцы (частотой 50 имп/с). Во всех случаях отмечено изменение резидуальной латенции, свидетельствующее о замедлении скорости проведения возбуждения по самым дистальным претерминальным…
Комплекс миастения — полимиозит
Патологическая утомляемость мышц является нередким симптомом всех форм полимиозита, однако у ряда больных наблюдали сочетание выраженных мышечных расстройств полимиозитического характера с несомненными клиническими и электрофизиологическими признаками, свидетельствующими о вовлечении в процесс синаптических структур, по характеру близкому патологическому процессу, наблюдаемому при миастении. Уже в конце XIX века Е. Wagner (1863, 1887) описал сочетание клиники полимиозита и…
Частично результаты обследования больных данной группы опубликованы в 1974 г. К настоящему времени под нашим наблюдением находятся 12 больных с этим клиническим синдромом. Все больные — девочки.
Начало заболевания отмечается в возрасте от 10 до 15 лет. При обследовании больных обращают на себя внимание выраженная мышечная гипотония, снижение, а иногда и выпадение сухожильных рефлексов.
Лишь…
Лечение при миастенических синдромах
В связи с неоднородностью механизмов развития нарушений нервно-мышечной передачи единого лечения миастенических синдромов нет. Воздействия на состояние нервно-мышечной передачи.
При большинстве форм миастенических синдромов в определенной степени оказываются эффективными антихолинэстеразные препараты — прозерин, оксазил, калимин и их аналоги (смотрите Лечение миастении).
Принципиально иной механизм действия другого препарата — гуанидина хлорида, способствующего выделению ацетилхолина из терминалей…
При обсуждении миастенического синдрома типа Ламберта — Итона следует отметить условность его названия, так как тщательное изучение клиники и механизмов развития данного заболевания позволило считать его неоднородным клиническим синдромом, обусловленным не только, как это предполагалось ранее, специфическим влиянием ракового процесса на нервно-мышечную передачу, но и типом реагирования нервно-мышечного синапса на целый ряд вредностей. Первыми миастенический…
Парестезии рук и ног наблюдаются у 50% больных. Все мужчины с синдромом Ламберта — Итона страдали импотенцией. Иллюстрациями к клинике миастенического синдрома Ламберта — Итона, связанного с бронхогенной мелкоклеточной карциномой, служат следующие наблюдения. Больной С., 43 лет, поступил в октябре 1975 г. с жалобами на слабость и утомляемость в мышцах ног и рук, мышцах туловища,…
Миастенический синдром при карциноматозных нейромиопатиях (патогенез)
При анализе патогенеза данного клинического синдрома обращает внимание относительная ясность механизмов, обусловливающих двигательные расстройства на синаптическом уровне, и полное отсутствие представления о причинах формирования синаптического дефекта.
Существенное место в раскрытии механизмов синаптических нарушений принадлежит изложенным выше результатам ЭМГ-ского исследования и результатам изучения ультраструктуры нервно-мышечных соединений.
Электрофизиологическое изучение больных с миастеническим синдромом типа Ламберта — Итона…
Источник: //www.meddr.ru/nervno-myshechnye_bolezni/
Читайте также[править | править код]
- Психофармакология Препараты для лечения депрессии и тревоги
- Антидепрессанты
- Лечение аффективных расстройств
- Препараты для лечения тревожных состояний
- Новые транквилизаторы
- Новые антидепрессанты
- Медикаментозное лечение психозов
Нервно-мышечные заболевания: что это такое, у детей, классификация НМЗ, все симптомы
Нервно-мышечные заболевания (НМЗ) – что это такое?
В медицине под этим термином подразумевается большая группа болезней, передающихся на генетическом уровне от родителей к ребенку.
Им характерно нарушение функций мускулатуры, низкая двигательная активность или ее отсутствие.
Такие последствия наступают в результате нарушений функций нервно-мышечных соединений, поражения мышц, спинномозговых нейронов или нервов.
Основные причины возникновения НМЗ – это аутоиммунные болезни, отравление различного рода веществами и наследственный фактор. Также сюда стоит отнести часть врожденных дефектов метаболизма.
Классификация
К НМЗ относятся:
- первичные прогрессирующие мышечные дистрофии (миопатии);
- вторичные прогрессирующие мышечные дистрофии;
- врожденные непрогрессирующие миопатии;
- миотонии;
- наследственные пароксизмальные миоплегии.
О каждом из видов поговорим подробнее дальше.
Первичные прогрессирующие мышечные дистрофии (миопатии)
К признакам прогрессирующих мышечных дистрофий (ПМД) относятся дегенеративные изменения в мышцах.
Они заключаются в истончении мускулатуры, замене мышечной ткани на жировую и соединительную.
Возникает фокальный некроз, поперечная исчерченность теряется.
Некоторые механизмы возникновения и течения болезни до сих пор не установлены. Причиной миопатии являются мембранные дефекты в мышечных клетках. Большие надежды на установление деталей патогенеза возлагают на микробиологов.
При миопатиях мышцы слабеют, затем атрофируются. Существует несколько форм этой болезни. Разные виды ПМД различают по типу наследования, срокам проявления, характеру и течению болезни, расположением атрофий.
Наследственные пароксизмальные миоплегии
Пароксизмальные миоплегии – редко встречающиеся наследственные заболевания у детей. Характеризуется периодическими приступами паралича мышц скелета. Наследуется по принципу, при котором для проявления болезни достаточно одного мутантного аллеля, локализованного в аутосоме.
Выделяют три формы:
- гиперкалиемическая;
- гипокалиемическая;
- нормокалиемическая.
Гипокалиемической формой пароксизмальной миоплегии чаще всего болеют мужчины.
Первые приступы заболевания появляются с 10 лет.
Приступ возникает в утренние или ночные часы. При этом чувствуется слабость в конечностях, шее, часто доходящая до паралича. В отдельных случаях паралич распространяется на лицевую мускулатуру и дыхательные пути.
Приступ сопровождается жаждой гиперемией, потливостью. Длительность может составлять от часа до семи дней. Женщины обычно испытывают приступ в первый день менструаций.
Гиперкалиемическую форму можно встретить реже, ее приступы начинаются в младшем возрасте. Приступ провоцируют холод и длительное бездействие мышц. Он начинается с расстройства чувствительности лицевых мышц, рук и ног.
Третья форма болезни проявляется до 10-летнего возраста. Приступы проходят в течение нескольких дней или недель. Они могут быть спровоцированы низкой температурой окружающей среды, употреблением алкоголя, большими физическими нагрузками.
Справка! Наследственные пароксизмальные миоплегии могут быть симптомами болезней щитовидной железы, Адиссона, реже встречаются при рвоте и поносе.
Врожденные непрогрессирующие миопатии
Непрогрессирующая миопатия характеризуется доброкачественностью, проявляется в младенчестве или в детстве. К этой разновидности НМЗ относятся болезни центрального стержня, намалиновые и митохондриальные миопатии. При этом возникают метаболические мышечные изменения. Пациент чувствует снижение сил, ослабление рефлексов.
Миотонии
Миотония – это замедленное расслабление мышцы после ее напряжения.
Болезнь разделяют на три вида:
- миотония действия;
- перкуссионная миотония;
- электромиографическая миотония.
Если больной с миотонией действия сожмет пальцы в кулак, а затем попытается быстро их разогнуть, то ему понадобится время, чтобы полностью выпрямить ладонь.
Вторая разновидность отличается сокращением мышц при интенсивном постукивании молоточком.
Чтобы обнаружить электромиографическую миотонию, потребуется игольчатый разряд. При этом аппарат показывает разряды высокой частоты, которые сначала учащенные, а потом снижают частоту.
Список всех НМЗ
- Болезни ЦС;
- Миотонии Томсена;
- Болезни Штейнерта-Куршманна;
- Миодистрофии Беккера;
- Миодистрофии Дюшенна;
- Миодистрофии Ландузи-Дежерина;
- Митохондриальные миопатии;
- Невральные амиотрофии Шарко-Мари-Тута;
- Немалиновые миопатии;
- Офталъмоплегические миопатии;
- Спинальные амиотрофии Верднига-Гоффманна;
- Спинальные амиотрофии Кугельберга-Веландер;
- Врожденная мышечная дистрофия;
- Мышечная дистрофия Беккера;
- Болезнь Помпе;
- Миотоническая дистрофия;
- Мышечная дистрофия Дюшенна.
Способы лечения
Лечение направлено на поддержку мышечных сил, замедление атрофирования.
Цель терапии заключается в том, чтобы пациент как можно дольше передвигался без посторонней помощи, т.к. в постоянном вертикальном положении возникают расстройства дыхания.
Основными способами лечения являются:
- Лечебно-физкультурный комплекс. Это различные активные и пассивные движения. Нельзя подвергать больного усиленной нагрузке. Упражнения должны быть регулярными;
- Ортопедические мероприятия. Применение специальных шин и проведение операций. Эти меры направлены на сохранение самостоятельных передвижений;
- Лекарственные препараты. Медикаменты, поддерживающие обмен веществ, устраняющие дефицит энергии и белка. Больным назначают фосфаден, преднизолон, нифедипин, витамин Е.
Неправильно поставленный предварительный диагноз приводит к ошибкам в диагностике формы НМЗ. Это увеличивает траты на осуществление дорогостоящих анализов и препятствует осуществлению профилактики рецидива, определения генетического статуса предков пациента и подбора адекватного патогенетического лечения, которым можно ограничиться в некоторых случаях.
по теме:
Источник: //doktor-ok.com/zabolevaniya/nervno-myshechnye
Симптомы невропатии бедренного нерва
Клинический симптомокомплекс бедренной невропатии зависит от топики процесса. При возникновении патологии на подвздошно-поясничном уровне развивается полный комплекс симптомов, включающий сенсорные, двигательные и вегетативно-трофические расстройства на всей иннервируемой бедренным нервом области. В редких случаях, при высоком разделении нерва, могут наблюдаться только сенсорные или только двигательные нарушения, иногда — мозаичная картина двигательных и чувствительных нарушений.
Полная невропатия бедренного нерва сопровождается лишь частичным нарушением работы подвздошно-поясничных мышц, благодаря существованию их альтернативной иннервации. Поэтому сгибание и супинация бедра практически не нарушены. Более выражен парез четырехглавой мышцы, отвечающей за разгибание ноги в коленном суставе. В связи с затруднительным разгибанием, пациенты стараются не сгибать ногу в колене. Затруднен бег и ходьба, особенно при необходимости подниматься по лестнице. Изменяется походка. Нога фиксирована в положении переразгибания. Наблюдается отсутствие коленного рефлекса.
К сенсорным нарушениям относятся расстройства тактильного и болевого восприятия на передне-внутренней поверхности бедра и голени, медиальном крае стопы. В этой же зоне наблюдаются трофические и вегетативные изменения, возможны ирритативные боли. В положении лежа на животе выявляются симптомы натяжения — боль по передней поверхности бедра при попытке максимально поднять прямую ногу (симптом Вассермана) или согнуть ногу в коленном суставе (симптом Мицкевича).
Невропатия бедренного нерва при его поражении в области паховой связки в общих чертах сходна с описанной выше клиникой. При высоком отхождении подкожного нерва могут наблюдаться преимущественно двигательные расстройства. Наряду с симптомами натяжения выявляется болезненность при надавливании посредине паховой связки.
Компрессия ствола бедренного нерва в канале Гунтера характеризуется болевой и тактильной гипестезией кожи медиального края коленного сустава, передне-внутренней поверхности голени и внутреннего края стопы. В этой же области наблюдаются парестезии и боли, которые усиливают свою интенсивность при разгибании голени. Последнее вынуждает пациента ходить и стоять, немного согнув ногу в колене. Коленный рефлекс не нарушен. Определяется болезненность в точке выхода подкожного нерва из приводящего канала, симптом Тинеля — появление парестезий по ходу нерва при его постукивании неврологическим молоточком.
Нервно-мышечные заболевания
Нервно-мышечные заболевания – условно выделяемая группа заболеваний, которые характеризуются нарушением функции мышц, прежде всего их слабостью.
Среди нервно-мышечных заболеваний выделяют болезни мышц, болезни периферических нервов, болезни нервно-мышечного соединения и болезни мотонейрона.
Один и тот же симптом слабости мышц может быть проявлением очень отличающихся по механизму заболеваний. Это определяет совершенно разный прогноз и способы лечения.
Приобретенные миопатии:
- воспалительные миопатии: (полимиозит, дерматомиозит, миозит с включениями, саркоидная миопатия;
- инфекционные миопатии (миопатии при ВИЧ, вирусные миозиты, бактериальные миозиты, паразитарные миозиты);
- лекарственные и токсические миопатии (кортикостероидная миопатия, миопатия при использовании лекарств для снижения холестерина, алкогольная миопатия, миопатия при критических состояниях).
- гипокалиемическая миопатия;
- гипофосфатемическая миопатия;
- миопатия при хронической почечной недостаточности;
- миопатия при диабете;
- миопатия при гипотиреозе;
- миопатия при гипертиреозе;
- миопатия при гиперпаратиреозе;
- болезнь Кушинга.
- миоглобинурия;
- каналопатии;
- наследственные миопатии;
- мышечные дистрофии.
Болезни периферических нервов
Болезни нервно-мышечного синапса
- Миастения
- Синдром Ламберта-Итона
- Ботулизм
- Клещевой паралич
Болезни мотонейрона
- Боковой амиотрофический склероз
- Болезни нижнего мотонейрона
- спинальная мышечная атрофия
- мономелический амиотрофический боковой склероз
- болезнь Кеннеди
- Болезни верхнего мотонейрона
- наследственный спастический парапарез
- первичный боковой склероз
Нервно-мышечное соединение или нервно-мышечный синапс – это соединение нервного окончания и мышечного волокна с образованием так называемой синаптической щели, в которой происходит передача импульса с нерва на мышечную мембрану.
Импульс передается при помощи нейромедиатора ацетилхолина, выделяемого окончанием нерва и прикрепляющегося затем к мышечной мембране.
При некоторых болезнях происходит нарушение нервно-мышечной передачи из-за недостаточного выделения ацетилхолина из нервного окончания или из-за нарушения прикрепления его к мембране мышечного волокна.
Миастения Гравис
Греческий термин миастения переводится как «мышечная слабость», а гравис как «серьезный». Миастения Гравис – это заболевание, характеризующееся выраженной мышечной слабостью и утомляемостью.
При миастении Гравис происходит нарушение передачи импульса с нервного волокна на мышечное.
В основе болезни лежит выработка аутоантител, которые блокируют присоединение нейромедиатора ацетилхолина к мышечной мембране в нервно-мышечном синапсе.
Как ставится диагноз?
Диагноз миастении ставится врачом на основе анализа крови и электронейромиографии. При необходимости назначается компьютерная томография грудной клетки для оценки размеров и состоянии вилочковой железы в качестве поиска возможной причины заболевания (выработка аутоантител).
Лечение
В лечении миастении Гравис используют антихолинэстеразные препараты (Пиридостигмин или Калимин) и препараты, подавляющие иммунную систему (преднизолон и другие). Удаление вилочковой железы (тимэктомия) выполняется, когда медикаментозная терапия малоэффективна. Также в лечении могут использоваться плазмаферез и иммуноглобулины.
Синдром Ламберта-Итона
Синдром Ламберта-Итона – это синдром мышечной слабости и утомляемости, развивающийся из-за аутоиммнунного процесса. Обычно причиной синдрома является злокачественный онкологический процесс, чаще всего, рак легких. Поэтому при постановке диагноза синдрома Ламберта-Итона пациенту всегда показано дообследование с целью онкопоиска.
Почему возникает синдром Ламберта-Итона?
Причиной являются антитела, вырабатываемые самим организмом(похожий аутоиммунный конфликт наблюдается и при миастении Гравис). В частности, антитела разрушают нервные окончания, нарушая тем самым регуляцию количества высвобождаемого нейромедиатора.
Когда количество нейромедиатора недостаточно, мышцы не могут сокращаться. Болезнь не является наследственной, страдают преимущественно молодые люди до 40 лет. Распространенность заболевания 1 на 1000000 человек.
У 40% больных синдромом Ламберта-Итона обнаруживают рак.
Как диагностируют синдром Ламберта-Итона?
Диагностика включает в себя анализ крови на антитела, тестовое введение антихолинэстеразного препарата, электронейромиографию.
Миастения
Заболеванию чаще всего подвержены женщины (2/3 от общего количества больных). Миастения имеет две формы – врождённую и приобретённую. При этом заболевании происходит нарушение передачи нервных импульсов, в результате чего возникает слабость в поперечнополосатых мышцах.
Заболевание связано с изменением функций нервно-мышечной системы. Слабеющие мышцы воздействуют на нормальную работу органов:у больного могут быть постоянно полузакрыты веки, происходит нарушение мочеиспускания, возникают трудности при жевании, ходьбе. В результате болезнь может привести к инвалидности и даже смерти.
Стадии и степени
Нервно-мышечные заболевания протекают по стадиям. Определить этап развития патологических процессов поможет врач невролог при помощи медицинской диагностики.
| Название | Описание |
| I стадия | Двигательные нарушения слабо выраженные. |
| II стадия | У больного присутствуют ярко выраженные клинические признаки и наблюдаются серьезные двигательные изменения. |
| III стадия | Пациент не может самостоятельно передвигаться. |
Нервно-мышечные заболевания миопатия Дюшенна на 2 этапе
Клиническая картина зависит от скорости развития патологических процессов и степени тяжести заболевания. Установить точный диагноз поможет врач невролог.