В список прогрессирующих мышечных дистрофий (ПМД) входят миодистрофии:
- псевдогипертрофическая Дюшенна;
- псевдогипертрофическая Беккера-Кинера;
- Эмери-Дрейфуса-Хогана;
- Роттауфа (фиброзирующая миопатия);
- ювенильная Эрба-Рота;
- окулярная (офтальмоплегия Грефе);
- плече-лопаточно-лицевая (Ландузи);
- окулофарингеальная;
- Дрейфуса;
- митохондриальные.
Клинические особенности
Симптомы заболевания проявляются до 5 лет. Самые ранние признаки мышечной дистрофии дюшенна включают задержку развития речи, ходьбы.
За этим следует постепенное ослабление и потеря мышц бедра и таза.
У пострадавшего есть проблемы, связанные с поднятием по лестнице со стула.
За определенное время наблюдается заметное утолщение мышц голени.
Узнать больше Все особенности синдрома Клиппеля Треноне Вебера
При прогрессивном вовлечении мышц груди, плеча, спины можно увидеть типичную позу.
Ребенок с трудом балансирует и постоянно падает. Вскоре, начинает ходить на носках благодаря сокращению тканей на пятках.
Лопатки, колени, бедра, позвоночник поражаются по мере прогрессирования заболевания, что приводит к сколиозу (искривлению позвоночника).
Пострадавший ограничен инвалидной коляской до 12 лет.
Хотя у большинства пациентов нормальный уровень интеллекта, признаки умственной отсталости могут наблюдаться в небольшом проценте случаев.
Мышцы сердца, легких также страдают, что приводит к смерти от респираторной или сердечной недостаточности.
Как только мышцы / респиратора диафрагмы становятся вовлеченными, пациенту становится трудно дышать, он зависит от аппарата для поддержки дыхания.
Прогноз при мышечных дистрофиях
Прогноз наследственных миопатий зависит от формы. Поскольку у большинства больных заболевание носит неуклонно прогрессирующий характер, то прогноз неблагоприятный.
Больные миодистрофией псевдогипертрофической Дюшенна обычно умирают на 3-м десятилетии жизни, чаще от прогрессирующей сердечной недостаточности. При псевдогипертрофической мышечной дистрофии Беккера-Кинера и мышечной дистрофии Эмери-Дрейфуса-Хогана больные обычно доживают до 40-60 лет. При миодистрофии Роттауфа-Мортъе-Бейера пациенты нередко доживают до 30-50 лет.
Статью подготовил и отредактировал: врач-хирург Пигович И.Б.
Видео:
Полезно:
Статьи по теме:
- Мышечные дистрофии у детей Мышечные дистрофии у детей — группа наследственных заболеваний, которая характеризуется прогрессирующей мышечной слабостью при отсутствии…
- Повышение уровня трансаминаз при заболеваниях мышц у детей Аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ) являются маркерами поражения печени, но в значительной степени эти ферменты…
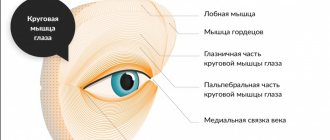
- Миопатия глаз Миопатия глаз (окулярная миопатия, окуло-фарингеальная миопатия) проявляется нарастающим на протяжении многих лет птозом и ограничением…
- Синдром Ламберта-Итона Синдром Ламберта-Итона обусловлен расстройством механизмов высвобождения ацетилхолина из пресинаптических пузырьков. Наблюдается у онкологических больных, чаще…
- Миопатии К миопатиям относят наследственные заболевания с прогрессирующей атрофией мышц, сопровождающейся нарастанием слабости в конечностях, В…
- Миопатии у детей Выделяют следующие формы врожденных миопатий у детей: болезнь «центрального стержня» (Шая-Межи); немалиновая миопатия; миотубулярная (центроядерная)…
Диагностика
Клиническое обследование
Клиническое обследование вызывает подозрение на МДД
Анализ креатинина фосфокиназы
Когда мышечные клетки умирают, пораженные выделяют фермент креатининфосфокиназу (СРК) в кровь. Таким образом, у пациентов с болезнью Дюшенна есть высокий уровень фермента в крови (в 20 раз больше верхнего предела нормы). Этот тест был единственным доступным методом обнаружения присутствия болезни до 1982 года.
Электромиография
Исследование, которое проверяет электрическую активность мышц. При дистрофии дюшена пораженные мышцы демонстрируют снижение электрической активности.
Мышечная биопсия
Испытание, при котором исследуется под микроскопом небольшая часть пораженной мышцы на наличие изменений.
Анализ дистрофина
Поскольку у пациентов с мышечной дистрофией дюшенна имеется дефектный ген, наблюдается нарушение в производстве белка дистрофина. Как правило, полный его дефицит наблюдается у 99% пострадавших. Анализ используется, чтобы отличить МДД от других форм дистрофии.
Эта форма диагностического инструмента дает точные результаты, поэтому может использоваться для установления диагноза.
Причины
Заболевание наследственное, сцепленное с X-хромосомой, поэтому болеют практически всегда мальчики. Девочки являются носителем патологического гена (мальчики редко доживают до половозрелого возраста, к тому же, как правило, стерильны). В хромосоме происходит изменение структуры гена, отвечающего за синтез белка дистрофина.
Хоть содержание дистрофина в скелетной мускулатуре предельно мало (тысячные доли процента), без него быстро развивается некроз мышечной ткани, развивается прогрессирующая дистрофия мышц. Если ген повреждается на участке, полностью разрушающем синтез белка-дистрофина, развивается дистрофия Дюшенна. При вовлечении в процесс малозначимых отделов белка, заболевание принимает форму дистрофии Беккера.
Лечение МДД
Несмотря на описание заболевания еще в 1868 году, нет надежного лечения мышечной дистрофии дюшенна.
Тем не менее, можно замедлить прогресс болезни и сохранить мышечную силу и совместную функцию, позволяя пациенту ходить и сидеть.
Узнать больше МДС, диагноз миелодиспластического синдрома: диагностика и лечение
Поскольку МДД поражает почти все мышцы тела, те, кто участвует в процессе лечения, сталкиваются с рядом проблем. Для эффективного управления инвалидизирующей болезнью необходим многодисциплинарный подход.
Применяются различные формы лечения, включающие легкие скобы, физиотерапию, легочную терапию (поддержку дыхания), хирургию.
Выбор конкретного лечения во многом зависит от клинического состояния пациента.
После идентификации гена исследователи разработали модели животных, которые имитируют болезнь. Такие клинические эксперименты улучшили ранее существовавшие знания о болезни.
Проводятся активные исследования, чтобы проверить, может ли инъекция нормальных мышечных клеток (от здоровых пациентов) или введение скорректированной формы гена дистрофина в мышечные клетки (генная терапия) отменять процесс заболевания.
Лечение мышечных дистрофий
Лечение первичных направлено на замедление темпа развития заболевания и максимальное сохранение способности больного к самообслуживанию.
Тактика лечения ПМД:
- диетотерапия с целью избежать развития у больного избыточной массы тела;
- ЛФК и двигательная активность, в частности, для поддержания функций опорных суставов и предотвращения контрактур;
- дыхательная гимнастика;
- медикаментозная терапия;
- социально-психологическая реабилитация;
- ортопедическая помощь.
При некоторых формах миодистрофий, в частности при миодистрофии Дюшенна, применение глюкокортикоидов иногда позволяет отодвигать наступление обездвиженности больного на годы. Поскольку такое лечение длительное и обычно сопровождается многими осложнениями, необходимо обдуманное назначение глюкокортикоидов. При ПМД применяют (с учетом возможных противопоказаний) преднизолон 1-2 мг/кг утром, через день, возможен и ежедневный прием преднизолона при мышечной дистрофии (утром однократно в дозе 0,75 мг/кг/сутки). Авторы предлагают применять также препарат этой же группы — дефлазакорт, обладающей меньшими побочными эффектами, приблизительно в той же дозе (по эффективности 6 мг дефлазакорта соответствуют 5 мг преднизолона). Длительность лечения зависит от эффективности и выраженности побочных действий препарата.
Показана симптоматическая и общеукрепляющая терапия. В частности, рекомендуют применение аденозина фосфата, трифосаденина, витамина Е, коферментов (например, кокарбоксилазы), негормональных анаболических средств (этилтиобензимидазола, оротовой кислоты), инозина, препаратов калия. При этом надо иметь в виду недоказанность объективных проявлений эффективности этих препаратов.
Рекомендуемая схема медикаментозного лечения ПМД:
Преднизолон внутрь (утром) по 1-2 мг/кг/сутки через день, длительность терапии определяют индивидуально, или Преднизолон внутрь (утром) по 0,75 мг/кг/сутки ежедневно, длительно или Дефлазакорт внутрь по 6 мг, длительно.
Методы диагностики заболевания Дюшенна-Беккера
Диагностикой болезни занимается невролог. Для подтверждения наличия патологии проводят ряд исследований:
- Тест ДНК. Позволяет с точностью определить наличие мутации гена.
- Биопсия мышечных тканей. Используется редко, заключается в заборе образца мышечных волокон и определение наличия дистрофина при помощи специального красителя.
- Пренатальное тестирование. Проводится на 11 неделе беременности с целью выявить риск нервно-мышечных нарушений, особенно если один или оба родителя являются носителями.
- Забор крови плода. Осуществляется на 18 неделе беременности, применяется в крайних случаях врачом-генетиком.
Внимание! Будущим родителям следует быть осторожными с тестами во время беременности. Однако если такая необходимость возникла, то предпочтение стоит отдавать раннему тестированию, чтобы минимизировать вероятность выкидыша.
Прогноз и профилактика
Для профилактики осложнений болезни пациент должен вести здоровый образ жизни, правильно питаться, достаточное время находиться на свежем воздухе. Запрещается ограничивать передвижения человека и держать его на постельном режиме. Без хотя бы минимальных попыток ходьбы мышцы разрушаются очень быстро.
Из-за того, что при дистрофии Дюшенна быстро повреждаются не только скелетные мышцы, но и сердце, органы дыхания, человек не может долго жить в таком состоянии. Даже при условии приема препаратов, облегчающих состояние, больные редко доживают до 35 лет. Больше 50% из них умирают еще в подростковом возрасте, без соответствующей помощи — еще быстрее.
При искусственной вентиляции легких, приеме поддерживающих лекарственных средств для сердца, использовании инвалидной коляски и трубки для кормления некоторые пациенты проживают 40-50 лет. Однако они нуждаются в постоянном уходе и наблюдении, так как не могут самостоятельно выполнить элементарные гигиенические процедуры и даже попросить о помощи.
Вылечить миодистрофию Дюшенна невозможно, однако облегчить состояние больного помогут лекарственные и ортопедические средства. Из-за значительных поражений внутренних органов пациенты обычно погибают в молодом возрасте. Но современные достижения в области применения стволовых клеток дают возможность надеяться на увеличение средней продолжительности жизни у людей, страдающих от этой болезни.
Лечение миопатии Дюшена
В настоящее время вылечить эту болезнь невозможно. Однако существует ряд способов, которые могут облегчить состояние пациента на разных стадиях развития миопатии и, возможно, несколько замедлить прогрессирование заболевание.
Ниже описаны способы лечения для определенных возрастных групп, но нередко при лечении одного пациента приходится сочетать сразу несколько методов терапии.
Пациенты дошкольного возраста
Обычно в этом возрасте детям с миопатией Дюшена еще не нужно лечение. Родителям могут предложить:
- Подробную информацию о миопатии Дюшена. Врач проведет с родителями беседу и подробно расскажет о том, как эта болезнь будет влиять на состояние ребенка, и какова ожидаемая продолжительность жизни пациента (большинство людей с таким диагнозом доживают лишь до 20-25 лет). При желании родители могут обратиться к другим специалистам (например, к психологу), а также в группы поддержки;
- Рекомендации относительно допустимых физических нагрузок для ребенка;
- Генетическую консультацию для членов семьи. Многие родители желают узнать, являются ли они носителями гена Дюшена. Это особенно важно для тех, кто в будущем планирует снова родить ребенка.
Уже в дошкольном возрасте ребенка начинают регулярно обследовать, чтобы, когда это будет необходимо, можно было вовремя начать лечение.
Пациенты в возрасте 5-8 лет
Детям такого возраста может потребоваться поддержка для мышц ног. Например, им могут рекомендовать надевать на ночь шины для лодыжки или более длинные шины, для голени.
Симптомы
Симптоматика проявляться начинает еще до трех лет. Ребенок начинает испытывать повышенную утомляемость, которая наступает достаточно быстро. Помимо этого отмечается:
- Отставание в развитии;
- Трудности с ходьбой и освоением данного навыка (часто ребенок начинает ходить на пальчиках);
- Попытка встать сопровождается активной помощью руками, что называют симптомом Говерса.
Последнее происходит из-за того, что мышцы ног перестают справляться с задаваемой на них нагрузкой.
Постепенно болезнь прогрессирует и к 12 годам пациент уже обычно нуждается в костылях. К периоду в 16-18 лет он уже прикован к инвалидному креслу.
Прогноз заболевания
На этот период приходятся следующие признаки миодистрофии Дюшенна:
- Неспособность не только вставать, но и самостоятельно садиться;
- Деформация скелета;
- Псевдогипертрофия икроножных мышц;
- Трудности с движением рук;
- Эндокринные расстройства;
- Кардиомиопатия и иные расстройства работы сердца;
- Ухудшение функционирования дыхательной мускулатуры;
- Реже – психические расстройства.
Если болезнь уже захватила сердце и легкие, то помочь такому пациенту уже нет возможности. То есть заболевание необратимо и ведет к летальному исходу в скором времени.
На фото симптомы миодистрофии
Терапия миодистрофии Дюшенна: старые и новые подходы
По словам Олсона, главное отличие новой стратегии с использованием вектора, вмещающего в себя компоненты для редактирования генома, от других терапевтических методов в том, что она устраняет причину болезни. А какие еще подходы разрабатывают ученые?
Рисунок 3. Животные модели миодистрофии Дюшенна. а — Проявления миодистрофии Дюшенна у мышей и собак. Вверху: у мышей mdx симптомы проявляются только в старости, и они склонны к образованию рабдомиосарком — опухолей мышечного происхождения. Размер мышей с нокаутами генов атрофина/дистрофина и интегрина/дистрофина значительно меньше, чем их ровесников дикого типа (BL10 и BL6). Внизу: проявления болезни у пятимесячной больной собаки. Различия между здоровой и больной двухлетними собаками. б — Сравнение продолжительности жизни здоровых и больных людей, собак и различных линий мышей.
[17]
Один из многообещающих подходов — это клеточная терапия. Хотя опыты с внутримышечной инъекцией миобластов от здоровых доноров провалились, технологии с использованием стволовых клеток и индуцированных плюрипотентных стволовых клеток (ИПСК) пока успешно испытываются на моделях не только миодистрофии Дюшенна, но и болезни Альцгеймера, Паркинсона, Хантингтона, спинальной мышечной атрофии, бокового амиотрофического склероза, аутизма и шизофрении [14–16]. Например, в 2013 году исследователи из Бостонской детской больницы (Boston Children’s Hospital’s Stem Cell Program) с помощью смеси трех малых молекул (форсколина, основного фактора роста фибробластов bFGF и ингибитора гликогенсинтазы киназы-3) перепрограммировали ИПСК из кожи пациентов с миодистрофией Дюшенна в мышечные клетки, которые затем успешно прижились у мышей. Сейчас из ИПСК получены кардиомиобласты и нейроны [2].
Другие исследования показывают, что восстановление нормального уровня синтеза оксида азота (NO), который снижается у больных из-за нарушения активности NO-синтазы (nNOS), ослабляет воспаление, повышает активность собственных стволовых клеток и реконструирует морфологию и функции скелетных мышц [3].
Уже в фазе II клинических испытаний находится препарат Givinostat — ингибитор гистондеацетилаз, который замедляет прогрессирование болезни в мышиной модели.
Такой массированный экспериментальный удар по миодистрофии Дюшенна вселяет надежду. Станет ли технология CRISPR/Cas9 ведущей в разработке терапии, которую смогут принять на вооружение клиницисты? Возможно, не за горами и публикации похожих работ по другим заболеваниям, где нужно избавиться от мутаций в одном-единственном гене? Это мы узнаем из ближайших выпусков Science (а также других почетных журналов).