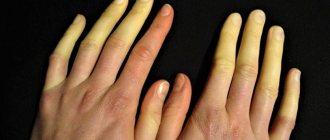
Симптомы
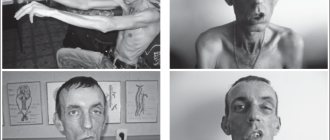
Классическая миотония Россолимо-Штейнерта-Куршмана дает о себе знать на 6-м году жизни и способна манифестировать до 35 лет. Чаще всего клинические признаки возникают в 10-20 лет. Они сочетают в себе типичные симптомы миопатии, поражения центральной нервной и сердечно-сосудистой систем, катаракты, эндокринных патологий.
Основные симптомы:
- Спазмы, особенно в сгибателях кисти, жевательных мышцах.
- Атрофические изменения в различных группах мышц.
- Поражаются дистальные отделы конечностей, височные мышцы, грудино-ключично-сосцевидные, височные, мимические.
- Миопатический парез гортани, сопровождающийся затрудненным дыханием и нарушениями голоса, снижение качества вентиляции легких, апноэ сна, аспирационная и застойная пневмония.
Примерно в половине случаев возможны патологии в сердечно-сосудистой системе:
- аритмия;
- блокада ножек пучка Гиса;
- гипертрофия левого желудочка.
Есть возможные симптомы поражения ЦНС:
- снижение интеллектуальных способностей;
- гиперсомния;
- легкая форма дебильности (в некоторых случаях).
Расстройства эндокринной системы поражают половую сферу.
У мужчин наблюдается:
- крипторхизм;
- импотенция;
- снижение либидо;
- гипогонадизм.
У женщин встречаются такие патологии:
- нарушение менструального цикла;
- гирсутизм;
- ранний климакс.
Также наблюдаются изменения в структуре волос, алопеция. Выпадение волос у мужчин наблюдается в области лба и на висках, у женщин – очаговое или диффузное облысение.
Врожденная форма заболевания проявляет себя еще во время внутриутробного развития. Плод демонстрирует пониженную двигательную активность. Для того чтобы это установить, нужно записаться на прием к акушеру-гинекологу, пройти акушерское УЗИ в третьем триместре.
После рождения у ребенка также проявляются признаки миопатии:
- Гипотония мышц диффузного типа: поражаются глазные, жевательные и мимические мышцы, дистальные отделы конечностей;
- Дыхательные расстройства.
Миотонические симптомы проявляются немного позже:
- олигофрения;
- задержка моторного развития.
Если симптомы прогрессируют быстро, это может привести к летальному исходу уже в детстве.
Миотония
Миотония (синоним болезнь Томсена) — это наследственное заболевание нервно-мышечной системы, характеризующееся своеобразными расстройствами движений в виде тонических мышечных спазмов, наступающих в начальной фазе активного движения. Заболевание передается по доминантному типу. Затрудняется расслабление мышцы после сильного сокращения ее в начале движения. Мышца остается сокращенной несколько секунд, затем медленно расслабляется. Последующие движения постепенно производить все легче, и спазмы совершенно прекращаются. Однако после отдыха, даже недлительного, тонический спазм мышц появляется с прежней силой. Спазмы захватывают мышцы конечностей, туловища и лица. Спазмы усиливаются при волнении и охлаждении. Первые симптомы миотонии развиваются в детском или юношеском возрасте и сохраняются у больных всю жизнь. Больные нередко отличаются атлетическим сложением, хорошо развитой мускулатурой. Прогноз благоприятен, но требуется правильный выбор специальности, не связанной с быстрыми двигательными реакциями,— противопоказана работа шофера, летчика, на конвейере и др.
Выделяют атрофическую миотонию, при которой наряду с миотоническими симптомами отмечаются эндокринные и дистрофические расстройства. У больных обнаруживаются атрофии мышц лица, шеи, предплечий и кистей, а также импотенция у мужчин, ранний климакс у женщин, ранняя катаракта, облысение, выпадение зубов, сухость кожи.
Лечение миотонии в основном симптоматическое. Назначение аскорбиновой кислоты и хинина (внутрь по 0,3—0,5 г 3—4 раза в день) временно снижает спазмы. Показаны препараты кальция и диета, бедная калием (ограничение картофеля и др.). Рекомендуются тепловые процедуры в виде световых и водных ванн, ионофорез с кальцием и хинином, массаж и лечебная физкультура.
См. также Наследственные болезни.
Миотония (myotonia; от греч. mys, myos — мышца и tonos — напряжение) — особое состояние мышц, выражающееся в том, что сократившаяся мышца долгое время не расслабляется, а затем расслабление происходит, но чрезвычайно медленно. Явление это свойственно произвольной поперечнополосатой мускулатуре, но может наблюдаться и в непроизвольной гладкой мускулатуре.
Миотония произвольной мускулатуры в чистом виде выражена при особом заболевании, которое носит название врожденной миотонией.
Врожденная миотония (myotonia congenita, болезнь Томсена) относится к наследственным заболеваниям нервно-мышечной системы, проявляется своеобразными расстройствами моторики — тоническими мышечными спазмами в начальной фазе активного движения.
Этиология недостаточно выяснена. Заболевание имеет наследственно-семейный характер, но часты и спорадические случаи; передается аутосомным геном, который наследуется доминантно.
Патогенез заболевания связан с наследственно обусловленными изменениями обмена в мышечной ткани. По-видимому, имеется нарушение ферментативных процессов, связанных с начальными фазами мышечного сокращения. В связи с положительным эффектом применения АКТГ при врожденной миотонии высказывается предположение о недостаточной функции надпочечников, приводящей к нарушению ионного равновесия калия.
Патологическая анатомия. В центральной и периферической нервной системе патологические изменения не выявлены. Отмечается увеличение поперечного размера мышечных волокон, сглаженность поперечной исчерченности, увеличение количества ядер сарколеммы.
Течение и симптомы. Первые признаки заболевания появляются в раннем детском или юношеском возрасте, усиливаются к 20 годам и сохраняются у больных всю жизнь.
Отмечается затруднение расслабления мышцы после сильного ее сокращения. Это происходит в начале произвольного движения: мышца, пришедшая в состояние тонического спазма, держится сокращенной несколько секунд, а затем медленно расслабляется. Последующие движения постепенно облегчаются и становятся нормальными. Однако после отдыха, даже не длительного, миотонический спазм мышц возобновляется с прежней интенсивностью. Ранним симптомом является расстройство походки. Особенно трудны первые шаги. Иногда при быстром движении больные теряют равновесие и падают. Тонические спазмы захватывают также мышцы рук, туловища и лица. Особенно затруднены движения в кисти и пальцах рук (например, сжимание и разжимание пальцев в кулак). Спазмы усиливаются при волнении и охлаждении и ослабевают от приема алкоголя. Мышечная сила у больных снижена по сравнению с нормой. Отмечается повышение механической и электрической возбудимости мышц при нормальной возбудимости нервов. При исследовании электровозбудимости обнаруживается замедленное расслабление мышцы, известное под названием «миотонической реакции». Отмечается изменение кожных и сухожильных рефлексов, имеющих также миотонический характер. Иногда наблюдается миотоническая реакция зрачков на конвергенцию и световое раздражение.
Прогноз благоприятен, но требует правильного выбора специальности больного, не связанной с быстрыми двигательными реакциями (работа на транспорте, конвейере и др.).
Лечение в основном симптоматическое. Назначают аскорбиновую кислоту и хинин внутрь (0,1—0,5 г 3—4 раза в день) или в виде внутримышечных инъекций (2 мл 50% раствора 1 раз в сутки) и тем самым временно снижают выраженность миотонического синдрома. Лечение большими дозами АКТГ (80—100 ЕД в сутки в течение 8—12 дней) обычно дает также нестойкий эффект. Показаны препараты кальция и диета, бедная калием. Рекомендуют тепловые процедуры в виде световых и водяных ванн, ионофорез с кальцием и хинином, массаж и умеренную лечебную физкультуру. См. также Наследственные болезни.
Миотония атрофическая (myotonia atrophica; синоним: дистрофическая миотония, миотоническая дистрофия, болезнь Куршмана — Баттена — Штейнерта) является, по-видимому, самостоятельным заболеванием, хотя не прекращаются попытки объединить ее с врожденной миотонией. Заболевание начинается в относительно более позднем возрасте, характеризуется неуклонно нарастающим течением и присоединением к миотоническим симптомам атрофических параличей.
Этиология. Заболевание передается по аутосомно-доминантному типу. Исследования хромосомного комплекса в культуре лейкоцитов периферической крови выявили лишнюю хромосому в группе малых акроцентриков (хромосом с дистально расположенной центромерой).
В патогенезе атрофической миотонии ведущую роль играет недостаточность функции надпочечников.
Течение и симптомы. Миотонический синдром особенно развит в мышцах пальцев и в жевательной мускулатуре, атрофические параличи локализуются в мышцах лица, плечевого пояса, а затем распространяются на конечности. Особенно характерным признаком является атрофия грудино-ключично-сосковых мышц. Выражены эндокринные расстройства в виде инфантилизма и бесплодия у женщин и нарушения половой функции у мужчин. Очень часто встречаются ранняя катаракта, атрофия кожи, облысение и др.
Прогноз: заболевание неуклонно прогрессирует и приводит к глубокой инвалидности.
Лечение такое же, как и при врожденной миотонии, но малоэффективно. См. также Парамиотония врожденная.
Диагностика
При проявлении вышеперечисленных симптомов необходимо обратиться за консультацией к врачу-неврологу. Чтобы подтвердить диагноз, проводится генеалогический анализ, анализ ДНК. Из диагностических процедур возможны:
- электромиография;
- исследование половых гормонов;
- ЭКГ;
- Электронейрография.
В некоторых случаях привлекаются врачи других специализаций: кардиологи, гинекологи, генетики, андрологи, эндокринологи.
Миотоническая дистрофия (болезнь Штейнерта, дистрофическая миотония)
Миотоническая дистрофия — аутосомно-доминантное многосистемное заболевание, характеризующееся сильно вариабельной экспрессией гена (клиническим полиморфизмом) у обоих полов по началу заболевания и тяжести течения. Главные клинические проявления: миотония, мышечная слабость, катаракты, аритмии сердца, облысение со лба, нарушенная толерантность к глюкозе, умственная отсталость. Мышечные судороги особенно выражены на руках, челюстях, языке (в виде фибрилляции). Одновременно отмечается постепенно усиливающаяся мышечная слабость в связи с дегенерацией отёчных мышечных клеток и атрофией волокон. Миотония и мышечная слабость у пациентов сочетаются с нарушением речи и глотания. Начальные признаки миотонической дистрофии варьируют. Миотония вначале выявляется только при специальном тестировании. Мышечные подёргивания и слабость обычно асимметричны. В первую очередь в патологический процесс вовлекаются лицевые и височные мышцы (статус миотонического лица), затем — шейные, плечевые, бедренные мышцы от проксимального направления к дистальному.
Наряду с нервно-мышечными симптомами при миотонической дистрофии отмечаются катаракты (очень ранний симптом), гипогонадизм (атрофия семенников), аменорея, дисменорея, кисты яичника, облысение со лба (особенно у мужчин), изменения проводимости сердца с аритмией, абдоминальные симптомы (на почве холелитиаза), прогрессирующая умственная отсталость.
Тяжесть клинических проявлений очень сильно различается даже в пределах одной семьи.
Миотоническая дистрофия характеризуется варьирующим началом заболевания: от пренатального периода до 50-60 лет. Различают 4 формы по возрастному «пику» начала заболевания: врождённая, юношеская, классическая (20-30 лет) и минимальная (50-60 лет). Это объясняется различиями в числе тринуклеотидных повторов в локусе миотонической дистрофии.
Смерть при миотонической дистрофии наступает в возрасте 50-60 лет (при классической форме) как следствие пневмонии, сердечных осложнений или других интеркуррентных заболеваний. Частота болезни, по-видимому, не различается в этносах и популяциях, хотя эффект родоначальника описан у канадцев французского происхождения. Обобщённо распространённость миотонической дистрофии можно оценить как 1:7500-1:10 000.
Генетика миотонической дистрофии хорошо изучена на генеалогическом, формально-генетическом и молекулярно-генетическом уровнях. У пациентов всех стран обнаружена одна и та же мутация в гене протеинкиназы мышечной дистрофии (символ гена DM-PK), локализованном в хромосоме 19ql3.3. Суть мутации — экспансия (увеличение числа) нестабильных CTG повторов в З’-нетранслируемой области гена. В норме число CTG повторов колеблется от 5 до 30. При миотонической дистрофии этот показатель значительно увеличивается и варьирует от 50 до 2500 и выше. Обнаружена корреляция между тяжестью течения и числом тринуклеотидных повторов. Чем больше повторов, тем раньше начинается заболевание и болезнь протекает тяжелее. Клиническая картина у гомозигот выражена в более тяжёлой форме. Во многих семьях с миотонической дистрофией в нескольких поколениях отмечается антиципация, т.е. более тяжёлая манифестация болезни и в более молодом возрасте в каждом последующем поколении. Этот признак описан для миотонической дистрофии давно и рассматривался в 40-х годах как статистический артефакт. Однако сведения о молекулярном дефекте свидетельствуют о возможности увеличения числа триплетов в поколениях. Описаны семьи более чем с тремя поколениями с миотонической дистрофией: в 1-м поколении — только катаракты, во 2-м поколении — умеренная слабость мышц, в 3-м поколении — врождённая форма. При миотонической дистрофии выражен импринтинг. У пациентов, рождённых больными матерями, имеется более тяжёлая форма болезни с более ранним началом, чем у пациентов, рождённых от больных отцов. Врождённая форма миотонической дистрофии наблюдается только при рождении детей больными матерями. Механизм импринтинга выяснен: экспансия триплетов происходит в мейозе у женщин, а при сперматогенезе этот процесс отсутствует. Уменьшение длины мутантного повтора (почти до нормы) у потомков с лёгкой клинической картиной и бессимптомным течением наблюдается при отцовской передаче гена. Одним из объяснений этого может быть селекция против длинных аллелей в мужском гаметогенезе. В некоторых семьях с миотонической дистрофией обнаружена нормальная длина тринуклеотидного сегмента гена. Это можно объяснить двумя вариантами. Во-первых, точковая мутация в гене миотонинпротеинкиназы может нарушать транскрипцию/трансляцию или стабильность мРНК, т.е. прерывать синтез этого фермента, что и вызывает миотоническую дистрофию. Во-вторых, имеются доказательства локусной генетической гетерогенности миотонической дистрофии. Обнаружен второй локус «классического» дистального фенотипа миотонической дистрофии, картированный на хромосоме 3q.
Статьи по теме: Генетика
Получить консультацию Специалиста
«Аксимед» — Медицинская клиника
- Информация
- Перейти на сайт
г. Киев, ул. Окипной Р.5
- (044) 390-00-55
- (067) 390-00-55
- (073) 390-00-55
- (099) 390-00-55
- Пн: 8.00 — 22.00
- Вт: 8.00 — 22.00
- Ср: 8.00 — 22.00
- Чт: 8.00 — 22.00
- Пт: 8.00 — 22.00
- Сб: 8.00 — 20.00
- Вс: 8.00 — 20.00
г. Киев, ул. Туманяна, 3
- (044) 390-00-55
- (067) 390-00-55
- (073) 390-00-55
- (099) 390-00-55
- Пн: 8.00 — 22.00
- Вт: 8.00 — 22.00
- Ср: 8.00 — 22.00
- Чт: 8.00 — 22.00
- Пт: 8.00 — 22.00
- Сб: 8.00 — 20.00
- Вс: 8.00 — 20.00
«Медиком» — Медицинская клиника
- Информация
- Перейти на сайт
г. Киев, пр. Г. Сталинграда, 6Д
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Пн: 8.00 — 20.00
- Вт: 8.00 — 20.00
- Ср: 8.00 — 20.00
- Чт: 8.00 — 20.00
- Пт: 8.00 — 20.00
- Сб: 8.00 — 20.00
- Вс: 8.00 — 20.00
г. Киев, ул. Борщаговская, 129/131
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Пн: 9.00 — 21.00
- Вт: 9.00 — 21.00
- Ср: 9.00 — 21.00
- Чт: 9.00 — 21.00
- Пт: 9.00 — 21.00
- Сб: 9.00 — 21.00
- Вс: Выходной
г. Киев, ул. В.Тютюнника (А. Барбюса), 37/1
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Пн: 8.00 — 21.00
- Вт: 8.00 — 21.00
- Ср: 8.00 — 21.00
- Чт: 8.00 — 21.00
- Пт: 8.00 — 21.00
- Сб: 8.00 — 21.00
- Вс: 8.00 — 20.00
г. Киев, ул.Кондратюка, 8
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Пн: 00:00 — 24:00
- Вт: 00:00 — 24:00
- Ср: 00:00 — 24:00
- Чт: 00:00 — 24:00
- Пт: 00:00 — 24:00
- Сб: 00:00 — 24:00
- Вс: 00:00 — 24:00
г.Киев, ул. Кондратюка, 8
- 1555
- (044) 503-77-77
- Пн: 00.00 — 24.00
- Вт: 00.00 — 24.00
- Ср: 00.00 — 24.00
- Чт: 00.00 — 24.00
- Пт: 00.00 — 24.00
- Сб: 00.00 — 24.00
- Вс: 00.00 — 24.00
«НоуХауМед» — Медицинский центр
- Информация
- Перейти на сайт
г. Киев, пр-т Героев Сталинграда 6, корпус 6
- (044) 233-50-40
- (067) 231-30-91
- (063) 831-50-80
- (095) 281-88-71(050) 311-39-05
- Пн: 9.00 — 19.00
- Вт: 9.00 — 19.00
- Ср: 9.00 — 19.00
- Чт: 9.00 — 19.00
- Пт: 9.00 — 19.00
- Сб: 9.00 — 17.00
- Вс: Выходной
г. Одесса, ул. Генуэзская, 24Б Аркадия (Le Silpo, KADORR City Mall).
- (048) 735-37-38
- (063) 390-28-70
- (096) 637-53-32(050) 311-39-05
- Пн: 9.00 — 19.00
- Вт: 9.00 — 19.00
- Ср: 9.00 — 19.00
- Чт: 9.00 — 19.00
- Пт: 9.00 — 19.00
- Сб: 9.00 — 17.00
- Вс: Выходной
Национальная детская специализированная больница «ОХМАТДЕТ»
- Информация
г. Киев, Стретенская,7/9 .
- (044) 236-69-42
- (044) 236-61-65
- (044) 236-69-47
- (044) 272-15-91
- (044) 272-02-34
- (044) 272-39-38
ПОЛЕЗНАЯ ИНФОРМАЦИЯ
- Аксиомы медицинской генетики
- Андреногенитальный синдром
- Болезни с аусомно-диминантным типом наследования
- Болезни с аутосомно-рецессивным типом наследования
- Болезни с Х-сцепленым доминантным типом наследования
- Генетическая классификация наследственных болезней
- Геномика
- Геномика патогенных бактерий и вирусов
- Значение генетики для медицины
- Клиническая диагностика наследственных болезней
- Микроцитогенетические синдромы
- Миодистрофия Дюкшена-Беккера
- Митохондриальная наследственность
- Мутации
- Синдром Эдвардса
- Синдром Дауна
Клинические проявления
В связи с варьированием начала заболевания в клинике различают следующие формы по возрастному принципу:
- врожденная форма — манифестация болезни начинается сразу после появления ребенка на свет;
- юношеский вариант — дебют миотонии в возрасте от одного года до периода полового созревания;
- классическая форма — начало клинических проявлений приходится на второй и третий десяток жизни;
- минимальный вариант — манифестация приходится на поздние сроки — шестой десяток жизни.
Характерно, что чем позднее проявляется болезнь, тем благоприятнее течение и лучше прогноз. Чаще всего встречается классическая форма болезни Штейнерта, для которой типичными являются следующие клинические симптомы:
- Миотония — проявляется спазмами жевательных мышц и сгибателей кистей рук, характерны атрофические изменения в разных группах мышц. Постепенно происходит угасание миотонических проявлений и прогрессирования мышечной дистрофии, внешне это выражается в печальной маске лица и отсутствии мимики. Опасным является парез мышц гортани с нарушением глотания, а также слабость дыхательной мускулатуры, в результате возможны приступы остановки дыхания во сне, развитие пневмонии.
- Сердечнососудистые нарушения — нарушения ритма сердца, гипертрофические изменения левого желудочка, выявляемые на ЭКГ, застойная сердечная недостаточность.
- Эндокринные расстройства (в основном затрагиваются половые функции) — уменьшение размеров половых органов, снижение сексуального влечения, у женщин — расстройства менструального цикла, ожирение.
- Общие изменения дистрофического характера — сухость и пигментация кожных покровов, выпадение частично или полностью волос и зубов, ранняя катаракта.
- Нарушения со стороны ЦНС — усталость, расстройства сна, апатия, потеря интеллекта.
Отдельно стоит отметить характерные клинические проявления врожденной формы дистрофической миопатии:
- уменьшение активных движений плода в утробе матери, выявляемое во время УЗИ;
- в период новорожденности — вялость, распространенная гипотония, особенно в жевательных, мимических, мышцах глазных яблок;
- сохранение и даже повышение сухожильных рефлексов;
- проблемы вскармливания, расстройства дыхания по типу респираторного дистресс-синдрома;
- задержка физического и нервно-психического развития, признаки олигофрении;
- стремительное прогрессирование заболевания, высокий риск внезапной смерти.
Медицинская помощь
Генетическое заболевание невозможно излечить полностью, поэтому целью лечения при болезни Россолимо-Штейнерта-Куршмана является купирование симптомов, улучшение общего состояния и социальная адаптация пациентов.
Принципы лечения заключаются в следующем:
- диета с низким содержанием солей калия (яблоки, спаржа, капуста, огурцы, виноград, зелень, кукуруза, ягоды, редис, мандарины, грейпфрут, лук, морковь, баклажаны, горох);
- исключение переохлаждений во избежание возникновения спазмов;
- применение препаратов хинина для стабилизации клеточных мембран, таких лекарств, как Дифенин, Новокаинамид, Диакарб — для снятия мышечных спазмов и уменьшения скованности, судорог, снижения внутричерепного давления;
- использование анаболических стероидов (Метанандростенолон, Ретаболил, Нерабол), витаминов группы В, АТФ для стимулирования мышечной массы;
- ЛФК, массаж, электромиостимуляция, ортопедические приспособления.
Перечисленные мероприятия дают хороший положительный эффект как при классической, так и при врожденной форме болезни. Полностью избавить больного от дистрофической миотонии они не могут, но продлить ему жизнь и улучшить ее качество способны.
Прогноз хуже у врожденной формы — летальность высока, дети могут не дожить до 3 лет. Юношеский вариант миотонии протекает достаточно тяжело и может привести уже в молодые годы к ограничению трудоспособности и ранней инвалидности.
В случае классической формы заболевание может протекать долго при проведении своевременных лечебно-коррекционных мероприятий. Наиболее благоприятный прогноз у поздно проявившихся форм болезни.
Профилактические мероприятия сводятся к тому, что женщине из семьи с неблагополучным анамнезом на стадии планирования беременности необходимо пройти обследование на наличие аномальных генов, ответственных за развитие мышечной дистрофии. Это целесообразно сделать также в случае наличия у родственников отца ребенка данной патологии.
Возможности к рождению деток должны решаться индивидуально в каждом конкретном случае врачами — генетиками после консилиума.
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — наследственное медленно прогрессирующее заболевание, в основе которого лежит дефект миотонин-протеинкиназы, приводящий к развитию миотонии в сочетании с дистрофическими изменениями мышечной ткани. Заболевание проявляется миотоническими спазмами, атрофическими изменениями мышц шеи, лица и дистальных отделов конечностей, снижением интеллекта, аритмиями и эндокринной патологией. Диагностика дистрофической миотонии основывается на клинических данных, результатах генеалогического анализа и исследования ДНК. Лечение симптоматическое, направленное против симптомов миотонии (фенитоин, прокаинамид, хинин, мочегонные) и мышечной дистрофии (анаболические стероиды, АТФ).
Этиологические причины
По результатам изучения генетического статуса заболевших выявлено: базисом патологии выступает дефект в гене DMPK, который находится в XIX хромосоме и ответственный за продукцию миотонин-протеинкиназы. Определяется большое учащение тринуклеотидных CTG-повторений в гене DMPK. Разновидность и степень тяжести формируется в соответствии с количеством повторений.
В нормальных условиях частота таких повторений колеблется от 10 до 35. Учащение их до 80 ведет к возникновению мягкого варианта патологического процесса. В случае же увеличения до 100-500 формируется поздний вариант болезни. Врожденная разновидность формируется при достижении 500-2000.
Изучения свидетельствуют, что учащаются повторения преимущественно в женских гаметах в мейотическом процессе. Поэтому в случае передачи патологии от матери у малыша формируется более тяжелая либо врожденная разновидности.
Врождённая форма
Второе название этой болезни – дистрофическая миотония 2 типа. Появиться она может сразу после рождения или даже во время внутриутробного развития плода. Однако нет никаких достоверных данных о том, что можно выявить болезнь по УЗИ. Чаще всего отмечается только замедленное шевеление плода, что может насторожить и стать причиной дальнейшего исследования.
После рождения явными становятся симптомы миопатии, что особенно выражено в мимической, жевательной и глазодвигательной мускулатуре. Появляются проблемы при глотании пищи и в дыхании. Проявляется задержка всех видов развития, а позднее диагностируется олигофрения. Быстрое прогрессирование симптомов приводит к смерти в раннем возрасте.
Диагностические критерии
Подозрение на болезнь Россолимо-Штейнерта — Куршмана может возникнуть у врача при наличии у пациента сочетания миотонических и дистрофических изменений в мышцах на фоне потери интеллекта и наличия сердечнососудистой и эндокринной патологии.
Полисистемность практически всегда свидетельствует о генетической природе заболевания. Такие больные подлежат анализу ДНК и проведению генеалогического анализа для подтверждения аутосомно-доминантного наследования патологии. В качестве информативных методов исследования используются электрокардиография, электронейромиография, анализы на гормоны.
В связи с многогранностью клинических проявлений к процессу постановки диагноза обычно привлекаются специалисты из разных отраслей медицины — генетики, кардиологии, эндокринологии, гинекологии, андрологии, неврологии.
Дифференциальный диагноз проводится между дистрофической миотонией и другими видами схожих заболеваний. В отличие от остальных для болезни Россолимо характерна мышечная атрофия. Нередко для подтверждения диагноза приходится прибегать к биопсии, чтобы определить уровень мышечного белка, который в тканях при данной патологии повышен.
Проводится также антенатальная диагностика методом исследования околоплодных вод.
Терапия
Лечение дистрофической миопатии не разработано, так как заболевание является генетическим. Пациентам показана диета с пониженным содержанием калия. Рекомендуется избегать переохлаждения, которое может провоцировать спазмы мышц.
Среди препаратов хорошим действием обладает хинин, прокаинамид, фенитоин, ацетазоламид. При необходимости могут быть назначены анаболические стероиды, АТФ, витамины группы В.
Прогноз дистрофической миотонии Штейнерта-Куршмана всегда неблагоприятный. Однако если симптомы незначительны, да и появились они в позднем возрасте, то прогноз для жизни является не таким угрожающим.
Общие сведения
Дистрофическая миотония Россолимо-Штейнерта-Куршмана является наследственным заболеванием и передается от родителей к детям по аутосомно-доминантному типу. Классическая форма этого заболевания развивается преимущественно в возрастном периоде от 10 до 20 лет. В более редких случаях встречается врожденная дистрофическая миотония Россолимо-Штейнерта-Куршмана, клинические симптомы которой проявляются сразу же после рождения.
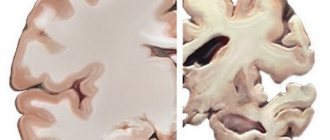
Морфологически при миотонии Россолимо-Штейнерта-Куршмана отмечается сочетание гипертрофических изменений одних мышечных волокон с атрофией других, замещение части мышечных волокон жировой и соединительной тканью. Изучение образцов мышечной ткани под электронным микроскопом показывает деструкцию миофибрилл и изменение размера митохондрий.
Открытие и суть заболевания
Россолимо, Штейнерт и Куршман изучали болезнь, являющуюся генетической патологией с аутосомно-доминантным типом наследования. Это значит, что один родитель имеет мутантный ген, больные дети при этом рождаются с вероятностью 50%. Заболевание носит характер семейного недуга и передается последующим поколениям по вертикали.
Сыновья и дочери в таких семьях болеют с одинаковой частотой, примерно 3 — 5 человек на 100 тысяч населения. Возраст начала заболевания, а также выраженность симптомов отличаются значительной вариабельностью.
Описаны ранние неонатальные и поздние формы, однако чаще всего заболевание дебютирует на втором, реже — на третьем десятке жизни. Отмечено, что передача болезни ребенку от матери является более прогностически неблагоприятной, чем от отца.
В основе болезни лежит дефект гена из 19 пары хромосом, который отвечает за синтез фермента миотонин-протеинкиназы. Это белок в норме присутствует не только в скелетной мускулатуре, но и в клетках миокарда и ЦНС.
Вот почему для дистрофической миотонии характерна полисистемность проявлений с поражением разных органов и систем. Неполноценность миотонин-протеинкиназы приводит к появлению мышечных спазмов вместе с атрофическими изменениями мускулатуры головы, шейного отдела, конечностей.
Наблюдается сочетание гипертрофии одних мышечных волокон с атрофией других и заменой их на жировую или соединительную ткани.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов — китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Диагностика миотонии Россолимо-Штейнерта-Куршмана
Типичное сочетание миотонии с признаками дистрофических изменений мышечной ткани, умственной отсталостью, нарушениями со стороны сердечно-сосудистой и эндокринной систем позволяет неврологупредположить миотонию Россолимо-Штейнерта-Куршмана. Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе наследования заболевания, и данные ДНК-анализа. Дополнительно проводится электромиография, электронейрография, исследования половых гормонов, ЭКГ. К диагностике пациентов с миотонией Россолимо-Штейнерта-Куршмана могут дополнительно привлекаться генетики, кардиологи, эндокринологи, гинекологи, андрологи.
При диагностике дистрофической миотонии ее необходимо дифференцировать ее от других видов миотонии. Так, наличие мышечных атрофий позволяет отличить миотонию Россолимо-Штейнерта-Куршмана от миотонии Томсена, для которой типична мышечная гипертрофия. От миотонии Беккера заболевание отличается ранним поражением мышц лица и доминантным типом наследования. Кроме того, следует проводить дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршман с миопатиями, БАС и амиотрофией Шарко-Мари-Тута.