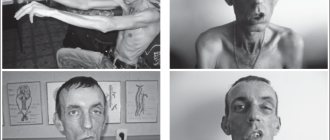
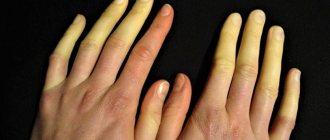
Symptoms
Classic Rossolimo-Steinert-Kurshman myotonia makes itself felt at the 6th year of life and can manifest itself up to 35 years of age.
Most often, clinical signs appear at 10-20 years of age. They combine typical symptoms of myopathy, damage to the central nervous and cardiovascular systems, cataracts, and endocrine pathologies. Main symptoms:
- Spasms, especially in the wrist flexors and masticatory muscles.
- Atrophic changes in various muscle groups.
- The distal parts of the limbs, temporal muscles, sternocleidomastoid, temporal, and facial muscles are affected.
- Myopathic laryngeal paresis, accompanied by difficulty breathing and voice disturbances, decreased quality of ventilation, sleep apnea, aspiration and congestive pneumonia.
In approximately half of the cases, pathologies in the cardiovascular system are possible:
- arrhythmia;
- bundle branch block;
- left ventricular hypertrophy.
There are possible symptoms of central nervous system damage:
- decreased intellectual abilities;
- hypersomnia;
- mild form of debility (in some cases).
Disorders of the endocrine system affect the genital area.
In men there is:
- cryptorchidism;
- impotence;
- decreased libido;
- hypogonadism.
The following pathologies occur in women:
- menstrual irregularities;
- hirsutism;
- early menopause
Changes in hair structure and alopecia are also observed. Hair loss in men is observed in the forehead and temples, in women - focal or diffuse baldness.
The congenital form of the disease manifests itself during intrauterine development. The fetus shows reduced motor activity. In order to establish this, you need to make an appointment with an obstetrician-gynecologist and undergo an obstetric ultrasound in the third trimester.
After birth, the child also shows signs of myopathy:
- Hypotonia of diffuse type muscles: the ocular, masticatory and facial muscles, distal parts of the limbs are affected;
- Respiratory disorders.
Myotonic symptoms appear a little later:
- oligophrenia;
- delayed motor development.
If symptoms progress quickly, it can be fatal in childhood.
Myotonia
Myotonia (synonymous with Thomsen's disease) is a hereditary disease of the neuromuscular system, characterized by peculiar movement disorders in the form of tonic muscle spasms that occur in the initial phase of active movement. The disease is transmitted by a dominant type. It becomes difficult to relax the muscle after a strong contraction at the beginning of the movement. The muscle remains contracted for a few seconds and then slowly relaxes. Subsequent movements gradually become easier and easier, and the spasms stop completely. However, after a rest, even a short one, tonic muscle spasm appears with the same force. Spasms involve the muscles of the limbs, torso and face. Spasms intensify with excitement and cooling. The first symptoms of myotonia develop in childhood or adolescence and persist in patients throughout their lives. Patients often have an athletic build and well-developed muscles. The prognosis is favorable, but the correct choice of a specialty that is not associated with fast motor reactions is required—work as a driver, pilot, on an assembly line, etc. is contraindicated.
Atrophic myotonia is distinguished, in which, along with myotonic symptoms, endocrine and dystrophic disorders are noted. Patients exhibit atrophy of the muscles of the face, neck, forearms and hands, as well as impotence in men, early menopause in women, early cataracts, baldness, tooth loss, dry skin.
Treatment of myotonia is mainly symptomatic. The administration of ascorbic acid and quinine (0.3-0.5 g orally 3-4 times a day) temporarily reduces spasms. Calcium supplements and a diet low in potassium (limitation of potatoes, etc.) are indicated. Thermal procedures in the form of light and water baths, iontophoresis with calcium and quinine, massage and physical therapy are recommended.
See also Hereditary diseases.
Myotonia (myotonia; from the Greek mys, myos - muscle and tonos - tension) is a special muscle condition, expressed in the fact that the contracted muscle does not relax for a long time, and then relaxation occurs, but extremely slowly. This phenomenon is characteristic of voluntary striated muscles, but can also be observed in involuntary smooth muscles.
Myotonia of voluntary muscles in its pure form is expressed in a special disease called congenital myotonia.
Congenital myotonia (myotonia congenita, Thomsen's disease) refers to hereditary diseases of the neuromuscular system, manifested by peculiar motor disorders - tonic muscle spasms in the initial phase of active movement.
The etiology is not well understood. The disease is hereditary and familial, but sporadic cases are also common; transmitted by an autosomal gene, which is inherited dominantly.
The pathogenesis of the disease is associated with hereditarily determined changes in metabolism in muscle tissue. Apparently, there is a violation of enzymatic processes associated with the initial phases of muscle contraction. In connection with the positive effect of the use of ACTH in congenital myotonia, it has been suggested that there is insufficient adrenal function, leading to an imbalance in the ionic balance of potassium.
Pathological anatomy. No pathological changes were detected in the central and peripheral nervous system. There is an increase in the transverse size of muscle fibers, smoothness of the transverse striations, and an increase in the number of sarcolemmal nuclei.
Course and symptoms. The first signs of the disease appear in early childhood or adolescence, intensify by the age of 20 and persist in patients throughout their lives.
There is difficulty in relaxing the muscle after a strong contraction. This occurs at the beginning of a voluntary movement: a muscle that has entered a state of tonic spasm remains contracted for several seconds and then slowly relaxes. Subsequent movements gradually ease and become normal. However, after rest, even not for a long time, myotonic muscle spasm resumes with the same intensity. An early symptom is gait disturbance. The first steps are especially difficult. Sometimes, when moving quickly, patients lose their balance and fall. Tonic spasms also affect the muscles of the arms, torso and face. Movements in the hand and fingers are especially difficult (for example, clenching and unclenching fingers into a fist). Spasms intensify with excitement and cooling and are weakened by drinking alcohol. Muscle strength in patients is reduced compared to the norm. There is an increase in mechanical and electrical excitability of muscles with normal excitability of nerves. When studying electrical excitability, a delayed relaxation of the muscle is detected, known as the “myotonic response”. There is a change in skin and tendon reflexes, which are also myotonic in nature. Sometimes a myotonic reaction of the pupils to convergence and light stimulation is observed.
The prognosis is favorable, but requires the correct choice of the patient’s specialty that is not associated with rapid motor reactions (work on transport, assembly lines, etc.).
Treatment is mainly symptomatic. Ascorbic acid and quinine are prescribed orally (0.1-0.5 g 3-4 times a day) or in the form of intramuscular injections (2 ml of a 50% solution once a day) and thereby temporarily reduce the severity of myotonic syndrome. Treatment with large doses of ACTH (80-100 units per day for 8-12 days) usually also produces an unstable effect. Calcium supplements and a diet low in potassium are indicated. Thermal procedures in the form of light and water baths, iontophoresis with calcium and quinine, massage and moderate physical therapy are recommended. See also Hereditary diseases.
Atrophic myotonia (myotonia atrophica; synonym: dystrophic myotonia, myotonic dystrophy, Kurshman-Batten-Steinert disease) is apparently an independent disease, although attempts to combine it with congenital myotonia continue. The disease begins at a relatively later age, is characterized by a steadily increasing course and the addition of atrophic paralysis to myotonic symptoms.
Etiology. The disease is transmitted in an autosomal dominant manner. Studies of the chromosomal complex in a culture of peripheral blood leukocytes revealed an extra chromosome in the group of small acrocentrics (chromosomes with a distal centromere).
In the pathogenesis of atrophic myotonia, adrenal insufficiency plays a leading role.
Course and symptoms. Myotonic syndrome is especially developed in the muscles of the fingers and masticatory muscles; atrophic paralysis is localized in the muscles of the face, shoulder girdle, and then spreads to the limbs. A particularly characteristic feature is atrophy of the sternocleidomastial muscles. Endocrine disorders are expressed in the form of infantilism and infertility in women and sexual dysfunction in men. Early cataracts, skin atrophy, baldness, etc. are very common.
Prognosis: the disease progresses steadily and leads to profound disability.
Treatment is the same as for congenital myotonia, but is ineffective. See also Paramyotonia congenital.
Diagnostics
If the above symptoms occur, you should consult a neurologist. To confirm the diagnosis, genealogical analysis and DNA analysis are performed. Diagnostic procedures are possible:
- electromyography;
- study of sex hormones;
- ECG;
- Electroneurography.
In some cases, doctors of other specializations are involved: cardiologists, gynecologists, geneticists, andrologists, endocrinologists.
Myotonic dystrophy (Steinert's disease, dystrophic myotonia)
Myotonic dystrophy is an autosomal dominant multisystem disease characterized by highly variable gene expression (clinical polymorphism) in both sexes in terms of disease onset and severity. Main clinical manifestations: myotonia, muscle weakness, cataracts, cardiac arrhythmias, baldness from the forehead, impaired glucose tolerance, mental retardation. Muscle cramps are especially pronounced in the arms, jaws, and tongue (in the form of fibrillation). At the same time, gradually increasing muscle weakness is noted due to degeneration of edematous muscle cells and fiber atrophy. Myotonia and muscle weakness in patients are combined with speech and swallowing disorders. The initial signs of myotonic dystrophy vary. Myotonia is initially detected only with special testing. Muscle twitching and weakness are usually asymmetrical. First of all, the facial and temporal muscles are involved in the pathological process (myotonic face status), then the cervical, shoulder, and femoral muscles from the proximal to the distal direction.
Along with neuromuscular symptoms in myotonic dystrophy, cataracts (a very early symptom), hypogonadism (testicular atrophy), amenorrhea, dysmenorrhea, ovarian cysts, baldness from the forehead (especially in men), changes in cardiac conductivity with arrhythmia, abdominal symptoms (due to cholelithiasis), progressive mental retardation.
The severity of clinical manifestations varies greatly, even within the same family.
Myotonic dystrophy is characterized by a variable onset of the disease: from the prenatal period to 50-60 years. There are 4 forms according to the age “peak” of the onset of the disease: congenital, juvenile, classic (20-30 years) and minimal (50-60 years). This is explained by differences in the number of trinucleotide repeats in the myotonic dystrophy locus.
Death with myotonic dystrophy occurs at the age of 50-60 years (with the classic form) as a result of pneumonia, cardiac complications or other intercurrent diseases. The incidence of the disease does not appear to vary across ethnic groups or populations, although a founder effect has been described in French Canadians. In general, the prevalence of myotonic dystrophy can be estimated as 1:7500-1:10,000.
The genetics of myotonic dystrophy has been well studied at the genealogical, formal genetic and molecular genetic levels. Patients from all countries were found to have the same mutation in the muscular dystrophy protein kinase gene (gene symbol DM-PK), located on chromosome 19ql3.3. The essence of the mutation is the expansion (increase in the number) of unstable CTG repeats in the 3'-untranslated region of the gene. Normally, the number of CTG repeats ranges from 5 to 30. With myotonic dystrophy, this figure increases significantly and varies from 50 to 2500 and above. A correlation was found between the severity of the disease and the number of trinucleotide repeats. The more repetitions, the earlier the disease begins and the more severe the disease. The clinical picture in homozygotes is more severe. In many families with myotonic dystrophy, anticipation is observed in several generations, i.e. more severe manifestation of the disease and at a younger age in each subsequent generation. This feature has been described for myotonic dystrophy for a long time and was considered in the 40s as a statistical artifact. However, information about the molecular defect indicates the possibility of increasing the number of triplets in generations. Families with more than three generations with myotonic dystrophy have been described: in the 1st generation - only cataracts, in the 2nd generation - moderate muscle weakness, in the 3rd generation - a congenital form. In myotonic dystrophy, imprinting is pronounced. Patients born to affected mothers have a more severe form of the disease with an earlier onset than patients born to affected fathers. The congenital form of myotonic dystrophy is observed only when children are born to sick mothers. The mechanism of imprinting has been clarified: expansion of triplets occurs in meiosis in women, but this process is absent during spermatogenesis. A decrease in the length of the mutant repeat (almost to normal) in offspring with a mild clinical picture and an asymptomatic course is observed during paternal transmission of the gene. One explanation for this could be selection against long alleles in male gametogenesis. Some families with myotonic dystrophy have a normal length of the trinucleotide gene segment. This can be explained in two ways. First, a point mutation in the myotonin protein kinase gene can impair transcription/translation or mRNA stability, i.e. interrupt the synthesis of this enzyme, which causes myotonic dystrophy. Second, there is evidence for locus genetic heterogeneity in myotonic dystrophy. A second locus for the “classic” distal myotonic dystrophy phenotype has been discovered, mapped to chromosome 3q.
Related articles: Genetics
Get advice from a Specialist
"Aximed" - Medical Clinic
- Information
- Go to website
Kyiv, st. Okipnoy R.5
- (044) 390-00-55
- (067) 390-00-55
- (073) 390-00-55
- (099) 390-00-55
- Mon: 8.00 – 22.00
- Tue: 8.00 – 22.00
- Wed: 8.00 – 22.00
- Thu: 8.00 – 22.00
- Fri: 8.00 – 22.00
- Sat: 8.00 – 20.00
- Sun: 8.00 – 20.00
Kyiv, st. Tumanyan, 3
- (044) 390-00-55
- (067) 390-00-55
- (073) 390-00-55
- (099) 390-00-55
- Mon: 8.00 – 22.00
- Tue: 8.00 – 22.00
- Wed: 8.00 – 22.00
- Thu: 8.00 – 22.00
- Fri: 8.00 – 22.00
- Sat: 8.00 – 20.00
- Sun: 8.00 – 20.00
"Medicom" - Medical Clinic
- Information
- Go to website
Kiev, G. Stalingrad Ave., 6D
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Mon: 8.00 – 20.00
- Tue: 8.00 – 20.00
- Wed: 8.00 – 20.00
- Thu: 8.00 – 20.00
- Fri: 8.00 – 20.00
- Sat: 8.00 – 20.00
- Sun: 8.00 – 20.00
Kyiv, st. Borshchagovskaya, 129/131
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Mon: 9.00 – 21.00
- Tue: 9.00 – 21.00
- Wed: 9.00 – 21.00
- Thu: 9.00 – 21.00
- Fri: 9.00 – 21.00
- Sat: 9.00 – 21.00
- Sun: Closed
Kyiv, st. V. Tyutyunnik (A. Barbussa), 37/1
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Mon: 8.00 – 21.00
- Tue: 8.00 – 21.00
- Wed: 8.00 – 21.00
- Thu: 8.00 – 21.00
- Fri: 8.00 – 21.00
- Sat: 8.00 – 21.00
- Sun: 8.00 – 20.00
Kyiv, Kondratyuk st., 8
- 1555
- (044) 503-00-00
- (044) 503-77-77
- Mon: 00:00 – 24:00
- Tue: 00:00 - 24:00
- Wed: 00:00 - 24:00
- Thu: 00:00 - 24:00
- Fri: 00:00 – 24:00
- Sat: 00:00 - 24:00
- Sun: 00:00 – 24:00
Kiev, st. Kondratyuka, 8
- 1555
- (044) 503-77-77
- Mon: 00.00 – 24.00
- Tue: 00.00 – 24.00
- Wed: 00.00 – 24.00
- Thu: 00.00 - 24.00
- Fri: 00.00 – 24.00
- Sat: 00.00 - 24.00
- Sun: 00.00 – 24.00
"KnowHowMed" - Medical Center
- Information
- Go to website
Kiev, Heroev Stalingrad Ave. 6, building 6
- (044) 233-50-40
- (067) 231-30-91
- (063) 831-50-80
- (095) 281-88-71(050) 311-39-05
- Mon: 9.00 – 19.00
- Tue: 9.00 – 19.00
- Wed: 9.00 – 19.00
- Thu: 9.00 – 19.00
- Fri: 9.00 – 19.00
- Sat: 9.00 – 17.00
- Sun: Closed
Odessa, st. Genuezskaya, 24B Arcadia (Le Silpo, KADORR City Mall).
- (048) 735-37-38
- (063) 390-28-70
- (096) 637-53-32(050) 311-39-05
- Mon: 9.00 – 19.00
- Tue: 9.00 – 19.00
- Wed: 9.00 – 19.00
- Thu: 9.00 – 19.00
- Fri: 9.00 – 19.00
- Sat: 9.00 – 17.00
- Sun: Closed
National Children's Specialized Hospital "OKHMATDET"
- Information
Kyiv, Stretenskaya, 7/9.
- (044) 236-69-42
- (044) 236-61-65
- (044) 236-69-47
- (044) 272-15-91
- (044) 272-02-34
- (044) 272-39-38
HELPFUL INFORMATION
- Axioms of medical genetics
- Andrenogenital syndrome
- Diseases with an ausomal dominant type of inheritance
- Diseases with autosomal recessive inheritance
- Diseases with X-linked dominant mode of inheritance
- Genetic classification of hereditary diseases
- Genomics
- Genomics of pathogenic bacteria and viruses
- The importance of genetics for medicine
- Clinical diagnosis of hereditary diseases
- Microcytogenetic syndromes
- Duchesne-Becker muscular dystrophy
- Mitochondrial inheritance
- Mutations
- Edwards syndrome
- Down syndrome
Clinical manifestations
Due to the varying onset of the disease, the clinic distinguishes the following forms according to age:
- congenital form - the manifestation of the disease begins immediately after the birth of the child;
- juvenile variant - the debut of myotonia between the ages of one year and puberty;
- classical form - the onset of clinical manifestations occurs in the second and third decades of life;
- the minimum option is that manifestation occurs late in life—the sixth decade of life.
It is typical that the later the disease manifests itself, the more favorable the course and better the prognosis. The most common form of Steinert disease is the classical one, for which the following clinical symptoms are typical:
- Myotonia - manifested by spasms of the masticatory muscles and flexors of the hands, characterized by atrophic changes in different muscle groups. Myotonic manifestations gradually fade away and muscular dystrophy progresses; outwardly this is expressed in a sad face mask and lack of facial expressions. Dangerous is paresis of the laryngeal muscles with impaired swallowing, as well as weakness of the respiratory muscles, which can result in attacks of respiratory arrest during sleep and the development of pneumonia.
- Cardiovascular disorders - heart rhythm disturbances, hypertrophic changes in the left ventricle detected on ECG, congestive heart failure.
- Endocrine disorders (mainly affecting sexual functions) - reduction in the size of the genital organs, decreased sexual desire, in women - menstrual cycle disorders, obesity.
- General changes of a dystrophic nature are dryness and pigmentation of the skin, partial or complete loss of hair and teeth, early cataracts.
- Central nervous system disorders - fatigue, sleep disorders, apathy, loss of intelligence.
It is especially worth noting the characteristic clinical manifestations of the congenital form of dystrophic myopathy:
- decrease in active movements of the fetus in the womb, detected during ultrasound;
- during the newborn period - lethargy, widespread hypotension, especially in the masticatory, facial, and eyeball muscles;
- preservation and even increase of tendon reflexes;
- feeding problems, breathing disorders such as respiratory distress syndrome;
- delayed physical and neuropsychic development, signs of mental retardation;
- rapid progression of the disease, high risk of sudden death.
Health care
The genetic disease cannot be cured completely, so the goal of treatment for Rossolimo-Steinert-Kurshman disease is to relieve symptoms, improve the general condition and social adaptation of patients.
The principles of treatment are as follows:
- a diet low in potassium salts (apples, asparagus, cabbage, cucumbers, grapes, herbs, corn, berries, radishes, tangerines, grapefruit, onions, carrots, eggplants, peas);
- avoiding hypothermia to avoid spasms;
- the use of quinine preparations to stabilize cell membranes, drugs such as Difenin, Novocainamide, Diacarb - to relieve muscle spasms and reduce stiffness, convulsions, and reduce intracranial pressure;
- the use of anabolic steroids (Methanandrostenolone, Retabolil, Nerabol), B vitamins, ATP to stimulate muscle mass;
- Exercise therapy, massage, electromyostimulation, orthopedic devices.
The listed measures give a good positive effect in both classical and congenital forms of the disease. They cannot completely rid the patient of dystrophic myotonia, but they can prolong his life and improve its quality.
The prognosis is worse for the congenital form - mortality is high, children may not live to see 3 years of age. The juvenile version of myotonia is quite severe and can lead to limited ability to work and early disability even in young years.
In the case of the classical form, the disease can last a long time if timely treatment and correction measures are carried out. The most favorable prognosis is for late-onset forms of the disease.
Preventive measures boil down to the fact that a woman from a family with an unfavorable history at the stage of pregnancy planning must undergo examination for the presence of abnormal genes responsible for the development of muscular dystrophy. It is advisable to do this also if the relatives of the child’s father have this pathology.
The possibility of having children should be decided individually in each specific case by geneticists after a consultation.
Dystrophic myotonia Rossolimo-Steinert-Kurshman is a hereditary slowly progressive disease, which is based on a defect in myotonin-protein kinase, leading to the development of myotonia in combination with dystrophic changes in muscle tissue. The disease is manifested by myotonic spasms, atrophic changes in the muscles of the neck, face and distal limbs, decreased intelligence, arrhythmias and endocrine pathology. Diagnosis of dystrophic myotonia is based on clinical data, the results of genealogical analysis and DNA research. Treatment is symptomatic, directed against the symptoms of myotonia (phenytoin, procainamide, quinine, diuretics) and muscular dystrophy (anabolic steroids, ATP).
Etiological reasons
Based on the results of studying the genetic status of the patients, it was revealed that the basis of the pathology is a defect in the DMPK gene, which is located on the XIX chromosome and is responsible for the production of myotonin-protein kinase. A large increase in trinucleotide CTG repeats in the DMPK gene is detected. The variety and severity are determined in accordance with the number of repetitions.
Under normal conditions, the frequency of such repetitions ranges from 10 to 35. Increasing their frequency to 80 leads to the emergence of a mild version of the pathological process. If it increases to 100-500, a late version of the disease is formed. The congenital variety is formed when it reaches 500-2000.
Studies indicate that repetitions increase predominantly in female gametes during the meiotic process. Therefore, in the case of transmission of pathology from the mother, the baby develops a more severe or congenital variety.
Congenital form
The second name for this disease is dystrophic myotonia type 2. It can appear immediately after birth or even during intrauterine development of the fetus. However, there is no reliable evidence that the disease can be detected by ultrasound. Most often, only slow fetal movement is noted, which can be alarming and cause further investigation.
After birth, symptoms of myopathy become obvious, which is especially pronounced in the facial, chewing and oculomotor muscles. Problems appear when swallowing food and breathing. All types of development are delayed, and later mental retardation is diagnosed. Rapid progression of symptoms leads to early death.
Diagnostic criteria
A doctor may suspect Rossolimo-Steinert-Kurshman disease if the patient has a combination of myotonic and dystrophic changes in the muscles against the background of loss of intelligence and the presence of cardiovascular and endocrine pathology.
Polysystemicity almost always indicates the genetic nature of the disease. Such patients are subject to DNA analysis and genealogical analysis to confirm the autosomal dominant inheritance of the pathology. Electrocardiography, electroneuromyography, and hormone tests are used as informative research methods.
Due to the versatility of clinical manifestations, specialists from various branches of medicine are usually involved in the diagnosis process - genetics, cardiology, endocrinology, gynecology, andrology, neurology.
Differential diagnosis is made between dystrophic myotonia and other types of similar diseases. Unlike the others, Rossolimo's disease is characterized by muscle atrophy. Often, to confirm the diagnosis, it is necessary to resort to a biopsy to determine the level of muscle protein, which is increased in the tissues of this pathology.
Antenatal diagnosis is also carried out by examining amniotic fluid.
Therapy
Treatment for dystrophic myopathy has not been developed, since the disease is genetic. Patients are prescribed a diet low in potassium. It is recommended to avoid hypothermia, which can provoke muscle spasms.
Among the drugs, quinine, procainamide, phenytoin, and acetazolamide have a good effect. If necessary, anabolic steroids, ATP, and B vitamins can be prescribed.
The prognosis of Steinert-Kurshman dystrophic myotonia is always unfavorable. However, if the symptoms are minor, and they appeared at a later age, then the prognosis for life is not so threatening.
General information
Dystrophic myotonia Rossolimo-Steinert-Kurshman is a hereditary disease and is transmitted from parents to children in an autosomal dominant manner. The classic form of this disease develops mainly in the age period from 10 to 20 years. In more rare cases, congenital dystrophic Rossolimo-Steinert-Kurshman myotonia occurs, the clinical symptoms of which appear immediately after birth.
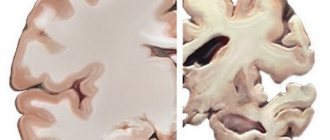
Morphologically, with Rossolimo-Steinert-Kurshman myotonia, there is a combination of hypertrophic changes in some muscle fibers with atrophy of others, replacement of some muscle fibers with adipose and connective tissue. Examination of muscle tissue samples under an electron microscope shows destruction of myofibrils and changes in mitochondrial size.
Discovery and essence of the disease
Rossolimo, Steinert and Kurshman studied the disease, which is a genetic pathology with an autosomal dominant mode of inheritance.
This means that one parent has a mutant gene, and sick children are born with a 50% probability. The disease is a family illness and is transmitted vertically to subsequent generations. Sons and daughters in such families get sick with the same frequency, approximately 3 - 5 people per 100 thousand population. The age at onset of the disease, as well as the severity of symptoms, vary widely.
Early neonatal and late forms have been described, but most often the disease debuts in the second, less often in the third decade of life. It has been noted that transmission of the disease to a child from the mother is more prognostically unfavorable than from the father.
The disease is based on a gene defect from the 19th pair of chromosomes, which is responsible for the synthesis of the enzyme myotonin-protein kinase. This protein is normally present not only in skeletal muscles, but also in myocardial and central nervous system cells.
That is why dystrophic myotonia is characterized by polysystemic manifestations affecting different organs and systems. Inferiority of myotonin-protein kinase leads to the appearance of muscle spasms along with atrophic changes in the muscles of the head, cervical region, and limbs.
There is a combination of hypertrophy of some muscle fibers with atrophy of others and their replacement with adipose or connective tissue.
Treatment
Treatment of muscular dystrophy is a complex and protracted process, however, currently no medicine has been created that completely heals the patient. All measures are aimed at making the patient’s life easier and restoring some lost abilities.
To slow down the development of the disease, the following drugs are prescribed:
- corticosteroids;
- vitamins B1;
- adenosine triphosphate (ATP).
In addition, to slow down the development process, fetal stem cells are used, which slow down the process of degeneration.
In addition, the following are prescribed as preventive measures:
- massage;
- physiotherapy;
- breathing exercises.
In addition to standard treatment options, it is important to constantly be guided by three main components in the process of life:
- Adequate physical activity.
- Timely psychological support.
- Dieting.
Physical activity
A person’s lack of desire to fight an illness has a negative impact on the body. Judge for yourself, passivity and reluctance to move depresses the already damaged muscular system. The muscles need to be given a load, since without load, degenerative processes begin to occur faster, thereby progressing at a faster pace.
Moderate physical activity and the use of supportive devices will be an excellent help in the fight against the disease.
If you have muscle pain, swimming, yoga, and stretching exercises are perfect.
Psychological support
Psychological support from the environment is important for a sick person. And if the illness is as serious as this one, even more so. For some, ordinary support from friends and family will be enough, while others may require qualified psychological help.
It is important to make it clear to such a person that he is not left alone with his problem. He must understand that he has someone to turn to, there are people who empathize and support him.
Diet
When it comes to diet and diet, there is a common belief that following an anti-inflammatory diet can slow the progression of the disease. This diet reduces inflammation, reduces glucose levels, removes toxins from the body and nourishes it with nutrients.
The essence of this diet is as follows:
- Refusal of foods containing “bad” fats and replacing them with “good” ones, introducing unsaturated fats into the diet, which are found in olive, flaxseed, sesame oil, and avocado.
- The use of meat and fish in the production of which did not use antibiotics or hormones.
- Complete removal of refined sugar and gluten from the diet.
- Eating the following foods - bok choy, broccoli, celery, pineapple, salmon, beets, cucumber, ginger, turmeric and other foods that have anti-inflammatory properties.
- Dairy products are only allowed based on sheep and goat milk.
- It is acceptable to drink herbal teas, lemonade, kvass, fruit drinks and natural juices.
Diagnosis of Rossolimo-Steinert-Kurshman myotonia
The typical combination of myotonia with signs of dystrophic changes in muscle tissue, mental retardation, disorders of the cardiovascular and endocrine systems allows the neurologist to assume Rossolimo-Steinert-Kurshman myotonia. The diagnosis is confirmed by the results of genealogical analysis, indicating an autosomal dominant type of inheritance of the disease, and DNA analysis data. Additionally, electromyography, electroneurography, sex hormone studies, and ECG are performed. Geneticists, cardiologists, endocrinologists, gynecologists, and andrologists can additionally be involved in the diagnosis of patients with Rossolimo-Steinert-Kurshman myotonia.
When diagnosing dystrophic myotonia, it is necessary to differentiate it from other types of myotonia. Thus, the presence of muscle atrophies makes it possible to distinguish Rossolimo-Steinert-Kurshman myotonia from Thomsen’s myotonia, for which muscle hypertrophy is typical. The disease differs from Becker's myotonia by early damage to the facial muscles and a dominant type of inheritance. In addition, a differential diagnosis of Rossolimo-Steinert-Kurshman myotonia should be made with myopathies, ALS and Charcot-Marie-Tooth amyotrophy.