Neuromuscular diseases: etymology, classification, symptoms and treatment
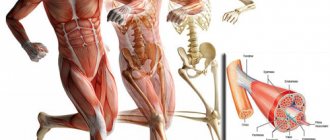
Neuromuscular diseases (NMDs) are one of the largest groups of hereditary diseases, characterized by dysfunction of voluntary muscles, decrease or loss of control of movements. The occurrence of these diseases is caused by a defect in embryonic development or a genetically determined pathology.
A characteristic manifestation of hereditary neuromuscular diseases is ataxia - a disorder of motor coordination and impaired motor skills. With static ataxia, balance is impaired while standing, with dynamic ataxia, coordination during movement is impaired.
The following symptoms are characteristic of neuromuscular diseases: weakness, muscle atrophy, spontaneous muscle twitching, spasms, numbness, etc.
If neuromuscular connections are disrupted, patients may experience drooping eyelids, double vision, and a number of other manifestations of muscle weakening, which only intensify during the day.
In some cases, disturbances in swallowing function and breathing are possible.
Classification of neuromuscular diseases
Neuromuscular diseases can be classified into four main groups depending on location:
- muscles;
- neuromuscular endings;
- peripheral nerves;
- motor neuron.
Based on the type and type of violations, they are divided into the following main groups:
- primary progressive muscular dystrophies (myopathies);
- secondary progressive muscular dystrophies;
- congenital non-progressive myopathies;
- myotonia;
- hereditary paroxysmal myoplegia.
Myopathy
The term myopathy (myodystrophy) unites a fairly large group of diseases that are united by a common feature: primary damage to muscle tissue. The development of myopathy can be triggered by various factors: heredity, viral infection, metabolic disorders and a number of others.
Inflammatory myopathies (myositis) include diseases caused by the inflammatory process. They develop as a result of autoimmune disorders and may be accompanied by other diseases of a similar nature. These are deramtomysitis, polymyositis, myositis with various inclusions.
Mitochondrial myopathies. The cause of the disease is structural or biochemical mitochondria. This type of disease includes:
- Kearns-Sayre syndrome;
- mitochondrial encephalomyopathy;
- myoclonus epilepsy with “torn red fibers.”
In addition to these diseases, there are a number of rare types of myopathies that affect the central core, endocrine system, etc.
When active, myopathy can lead to disability and further immobilization of the patient.
Secondary progressive muscular dystrophies
The disease is associated with disruption of the functioning of peripheral nerves, disruption of the supply of organs and tissues with nerve cells. As a result, muscle dystrophy occurs.
There are three types of secondary progressive muscular dystrophy: congenital, early childhood and late. In each case, the disease proceeds with a greater or lesser degree of aggression. For people with this diagnosis, the average life expectancy is from 9 to 30 years.
Congenital non-progressive myopathies
These include hereditary non-progressive or weakly progressive muscle diseases diagnosed in the prenatal period or immediately after the birth of the baby. The main symptom is muscle hypotonia with pronounced weakness. This disease is otherwise called “flaccid child syndrome,” which accurately characterizes its condition.
In most cases, the area of the lower extremities is affected, less often - the upper extremities; in exceptional cases, damage to the cranial muscles occurs - impaired facial expressions and eye movements.
During the development and growth of a child, problems with motor skills are noted; children often fall, begin to sit and walk late, and cannot run or jump. There are no intellectual impairments. Unfortunately, this type of myopathy is incurable.
Symptoms
For all types of myopathies, the main symptom is muscle weakness. Most often, the muscles of the shoulder girdle, hips, pelvic area, and shoulders are affected.
Each type is characterized by damage to a specific muscle group, which is important to consider when diagnosing.
The defeat occurs symmetrically, so he is able to carry out actions in stages, gradually including various areas in the work.
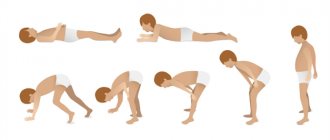
If the legs and pelvic area are affected, in order to get up from the floor, you must first lean your hands on the floor, kneel down, grab a support, and after that the patient can sit on a chair or bed. He will not be able to stand up on his own without using his hands.
With myopathy, cases of damage to the facial muscles are the least common. This is ptosis (drooping of the upper eyelid), the upper lip droops. Speech problems arise due to impaired articulation, and swallowing function may be impaired.
Most myopathies occur with almost identical symptoms. Over time, muscle tissue atrophies, against the background of which connective tissue actively grows.
Visually it looks like trained muscles - the so-called. pseudohypertrophy. In the joints themselves, contractures form and the muscle-tendon fiber tightens.
As a result, pain appears and joint mobility is limited.
Myoplegia
Like myopathies, these are hereditary neuromuscular diseases characterized by attacks of muscle weakness or paralysis of the limbs. The following types of myoplegia are distinguished:
- hypokalemic;
- hyperkalemic;
- normokalemic.
An attack of myoplegia is caused by a redistribution of potassium in the body - there is a sharp decrease in it in the intercellular fluid and plasma, and an increase (excess) in the cells.
In muscle cells, membrane polarization is disrupted and the electrolytic properties of muscles change.
During an attack, the patient experiences severe weakness of the limbs or torso, manifestations in the pharynx, larynx, and effects on the respiratory tract are possible. This can be fatal.
Myasthenia gravis
The disease most often affects women (2/3 of the total number of patients). Myasthenia has two forms - congenital and acquired. With this disease, the transmission of nerve impulses is disrupted, resulting in weakness in the striated muscles.
The disease is associated with changes in the functions of the neuromuscular system. Weakening muscles affect the normal functioning of organs: the patient may have constantly half-closed eyelids, difficulty urinating, and difficulties in chewing and walking. As a result, the disease can lead to disability and even death.
Motor Neurone Diseases (MND)
Motor neuron diseases are characterized by damage to the motor neurons of the brain and spinal cord. The gradual death of cells affects muscle function: they gradually weaken, and the affected area increases.
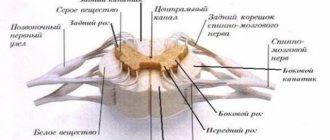
The brain neurons responsible for movement are located in the cerebral cortex. Their branches - axons - descend into the spinal cord, where contact occurs with the neurons of this department. This process is called synapse.
As a result, a neuron in the brain releases a special chemical (transmitter) that transmits a signal to neurons in the spinal cord.
These signals are responsible for the contraction of muscles in various parts: cervical, thoracic, bulbar, lumbar.
Depending on the severity of neuronal damage and their localization, several types of BND are distinguished. In many ways, the manifestations of the diseases are the same, but as the disease progresses, the difference becomes more and more significant.
There are several different types of BND:
Amyotrophic lateral sclerosis
It is one of the four main types of motor neuron disease. It occurs in 85% of patients diagnosed with motor neuron disease. The affected area can be both neurons of the brain and spinal cord. As a result, muscle atrophy and spasticity occur.
With ALS, there is weakness and increasing fatigue in the limbs. Some people have weakness in their legs when walking and weakness in their arms, making it difficult to hold things in their hands.
In most cases, the disease is diagnosed before the age of 40, and the disease does not affect the intellect at all. The prognosis for a patient diagnosed with ALS is not the most favorable - from 2 to 5 years. But there are exceptions: the most famous person of all, who lived with this diagnosis for more than 50 years, is Professor Stephen Hawking.
Progressive bulbar palsy
Associated with speech and swallowing disorders. The prognosis from the date of diagnosis is up to three years from the date of diagnosis;
Primary literal sclerosis
Affects only the neurons of the brain and affects the lower extremities. In rare cases, it is accompanied by impaired hand movements or speech problems. In later stages it can progress to ALS.
Progressive muscular atrophy
Occurs when the motor neurons of the spinal cord are damaged. The first manifestations are expressed in hand weakness. The prognosis for this disease is from 5 to 10 years.
Diagnostics
To establish an accurate diagnosis, it is important to conduct the following studies:
- biochemical. Determination of muscle enzymes, primarily creatine phosphokinase (CPK). The level of myoglobin and aldolase is determined;
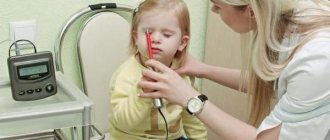
- electrophysiological. Electromyography (EMG) and electroneuromyography (ENMG) help in differentiating between primary and secondary myopathy. They also help to identify whether the spinal cord or peripheral nerve is primarily affected;
- pathomorphological. Consists of performing a muscle biopsy. Studying the material also helps to differentiate primary or secondary myopathy. Determining the level of dystrophin makes it possible to distinguish Duchenne myopathy from Becker myodystrophy, which is important for prescribing the correct treatment;
- DNA diagnostics. The study of DNA leukocytes allows us to identify hereditary diseases in 70% of patients.
Treatment of neuromuscular diseases
When one of the diagnoses related to neuromuscular diseases is made, in each specific case the treatment is selected individually, taking into account all the tests obtained. The patient and his relatives must initially understand that this is a long and very complex process that requires large financial costs.
Difficulties in prescribing treatment are also associated with the fact that it is not always possible to accurately determine the primary metabolic defect. At the same time, the disease is constantly progressing, which means that treatment should primarily be aimed at slowing the progression of the disease. This will help preserve the patient’s ability to self-care and affect his quality of life.
Treatment methods for neuromuscular diseases
- Correction of skeletal muscle metabolism. Drugs that stimulate metabolism, potassium supplements, vitamin complexes, and anabolic steroids are prescribed;
- Stimulation of the segmental apparatus. Neurostimulation, myostimulation, reflexology, balneotherapy, physical therapy (exercises and load are selected individually);
- Correction of blood flow. Various types of massage, thermal procedures on certain areas, oxygen barotherapy;
- Diet and parenteral nutrition to provide the body with all the necessary nutrients - protein, potassium salts, vitamins of the required group;
- Corrective sessions with an orthopedist. Correction of contractures, deformities of the chest and spine, etc.
Today, no medicine has been invented that can make any person absolutely healthy in an instant. Despite the complexity of the situation, it is important for a patient with a neuromuscular disease to continue to live the best possible quality of life. The example of Hawking, who was confined to a wheelchair for more than 50 years but continued to conduct research, suggests that illness is not a reason to give up.
Source: //onevrologii.ru/nervno-myshechnye-patologii/nervno-myshechnye-zabolevaniya
Congenital non-progressive myopathies
These include hereditary non-progressive or weakly progressive muscle diseases diagnosed in the prenatal period or immediately after the birth of the baby. The main symptom is muscle hypotonia with pronounced weakness. This disease is otherwise called “flaccid child syndrome,” which accurately characterizes its condition.
In most cases, the area of the lower extremities is affected, less often - the upper extremities; in exceptional cases, damage to the cranial muscles occurs - impaired facial expressions and eye movements.
Neuromuscular diseases – Med Dr
Neuromuscular diseases are the largest group of all diseases of the nervous system. These include various types of myopathies, neural and spinal amyotrophies, myasthenia gravis, myotonia and periodic paralysis.
The relatively high frequency of these diseases, severe disability in most of them, and damage in childhood or prime age make them very relevant. Meanwhile, practicing doctors are not fully familiar with this problem, especially with advances in the study of pathogenesis, modern diagnostic methods and new treatment methods.
The monograph presented to the attention of readers was created by employees of two teams who have been studying various aspects of muscle pathology for more than a quarter of a century; The beginning of this study was laid by N. I. Grashchenkov, Z. L. Lurie, L. B. Perelman. This was reflected in the construction of the monograph and the choice of materials.
Based on the relatively small volume of the book, the authors focused on those aspects of the problem in the development of which they were directly involved and have the greatest experience.
Due to the fact that the book is essentially a collective monograph, the creation of which was attended by authors belonging to two scientific directions - clinical-genetic, based mainly on the use of biochemical methods of analysis, and clinical-pathophysiological, using as an analysis tool in mainly electromyographic research methods - when presenting individual problems, the authors were unable to completely avoid contradictions in some issues. However, they did not strive for a monotonous presentation, leaving the reader the opportunity to create his own opinion after familiarizing himself with the factual material.
The book is not a textbook or reference guide on clinical myology, so it is in vain to look in it for an answer to all the readers’ questions. Thus, based on the availability of available publications, the authors do not consider amyotrophic lateral sclerosis, spinal amyotrophies, hereditary lesions of the peripheral nervous system; other sections are presented more fully.
Along with a discussion of the problems of progressive muscular dystrophies, non-progressive muscular dystrophies are described, as well as neuromuscular disorders that arise from diseases of the endocrine and metabolic glands. Practice shows that motor dysfunction long precedes other symptoms of endocrine suffering.
The clinical issues, diagnosis and treatment of inflammatory and autoimmune diseases of the neuromuscular system are covered. Early diagnosis of these diseases is especially important, as it provides early therapy that can radically change the course and prognosis of the disease.
In this regard, treatment is described in more detail in these sections.
The presentation of particular problems is preceded by two chapters that have both theoretical and purely practical significance: on the role of biochemical research methods and clinical electromyography in the study of the mechanisms of development and diagnosis of neuromuscular diseases.
We believe that the inclusion of these chapters, written in relation to the problems of clinical myology, will not only help the reader in understanding the problems raised in the monograph, but will also contribute to the wider use of these methods in the diagnosis and monitoring of pathological processes.
The implementation of the research on which the book is based required the participation of a large number of employees of those institutions where these studies were carried out - the Institute of General Pathology and Pathological Physiology of the USSR Academy of Medical Sciences, the Clinic of Nervous Diseases and Faculty Surgery of the I. M. Sechenov Moscow Medical Institute, the Central Clinical Hospital No. 3 MPS. The authors express their heartfelt gratitude to the staff and managers of these institutions, as well as to everyone who contributed to the creation of the book with advice and critical comments.
“Neuromuscular diseases”, B.M. Gekht, N.A. Ilyina
Badalyan L. O. Clinical genetic analysis and issues of classification of progressive muscular dystrophies. — In the book: Issues of clinical neurogenetics. M., 1973, p. 68 - 79. Bondarenko E. S.
Hereditary muscular dystrophies/CIU. - M., 1976. Gadzhiev S. A., Logel L. V., Vanevsky V. L. Diagnosis and surgical treatment of myasthenia gravis. - L.: Medicine, 1971. Gausmanova-Petrusevich I.
Muscular diseases/Trans. With…
Adams R. Diseases of muscle: A stucfy in pathology. 3 ed. — 1975. Bradley WG Disorders of peripheral nerves. — Oxford; London, 1974. Buchthal F. An introduction to electromyography. - Copenhagen, 1957. Dubowitz V., Brooke MH Muscle biopsy: A modern approach. - London, Philadelphia, Toronto, 1973. Emeryk V., Strugalska M. Evaluation of results...
Myasthenic syndrome with terminal polyneuropathy
A special form of myasthenic disorders caused by damage to the terminal branches of motor nerves and gross disorders of neuromuscular transmission is described by us in 1979.
terminal polyneuropathy with myasthenic syndrome. To date, we have been observing 13 patients with this symptom complex (12 men and 1 woman) for 12 years.
In 2 patients, the disease began at the age of over 30 years,...
First of all, with this disease, typical EMG changes are observed, indicating gross changes in neuromuscular transmission, a decrease in the amplitude of the evoked muscle action potential, a gross block of neuromuscular transmission during stimulation with rare frequencies (1 and 3 pulses/s) and tetanization of the muscle (frequency 50 imp/s). In all cases, a change in residual latency was noted, indicating a slowdown in the speed of excitation along the most distal preterminal...
Myasthenia gravis-polymyositis complex
Pathological muscle fatigue is a common symptom of all forms of polymyositis, however, in a number of patients, a combination of pronounced muscle disorders of a polymyositic nature with undoubted clinical and electrophysiological signs indicating the involvement of synaptic structures in the process was observed, similar in nature to the pathological process observed in myasthenia gravis. Already at the end of the 19th century, E. Wagner (1863, 1887) described the combination of the clinic of polymyositis and ...
Partial results of examination of patients in this group were published in 1974. To date, 12 patients with this clinical syndrome are under our supervision. All patients are girls.
The onset of the disease is observed between the ages of 10 and 15 years. When examining patients, attention is drawn to pronounced muscle hypotonia, decrease, and sometimes loss of tendon reflexes.
Only…
Treatment for myasthenic syndromes
Due to the heterogeneity of the mechanisms of development of neuromuscular transmission disorders, there is no uniform treatment for myasthenic syndromes. Impact on the state of neuromuscular transmission.
In most forms of myasthenic syndromes, anticholinesterase drugs - prozerin, oxazil, kalimine and their analogues - are effective to a certain extent (see Treatment of myasthenia gravis).
A fundamentally different mechanism of action of another drug - guanidine chloride, which promotes the release of acetylcholine from the terminals...
When discussing myasthenic syndrome of the Lambert-Eaton type, it should be noted that its name is conventional, since a thorough study of the clinical picture and mechanisms of development of this disease made it possible to consider it a heterogeneous clinical syndrome, caused not only, as previously assumed, by the specific influence of the cancer process on neuromuscular transmission, but also by the type of response of the neuromuscular synapse to a number of hazards. The first myasthenic...
Paresthesia of the arms and legs is observed in 50% of patients. All men with Lambert-Eaton syndrome suffered from impotence. The following observations illustrate the clinical picture of Lambert-Eaton myasthenic syndrome associated with bronchogenic small cell carcinoma. Patient S., 43 years old, was admitted in October 1975 with complaints of weakness and fatigue in the muscles of the legs and arms, muscles of the trunk,…
Myasthenic syndrome in carcinomatous neuromyopathies (pathogenesis)
When analyzing the pathogenesis of this clinical syndrome, attention is drawn to the relative clarity of the mechanisms causing movement disorders at the synaptic level and the complete lack of understanding about the reasons for the formation of the synaptic defect.
A significant place in revealing the mechanisms of synaptic disorders belongs to the results of the EMG study outlined above and the results of studying the ultrastructure of neuromuscular junctions.
Electrophysiological study of patients with myasthenic syndrome of the Lambert-Eaton type...
Source: //www.meddr.ru/nervno-myshechnye_bolezni/
Read also[edit | edit code]
- Psychopharmacology Drugs for the treatment of depression and anxiety
- Antidepressants
- Treatment of mood disorders
- Medicines to treat anxiety
- New tranquilizers
- New antidepressants
- Drug treatment of psychosis
Neuromuscular diseases: what is it, in children, classification of neuromuscular diseases, all symptoms
Neuromuscular diseases (NMDs) – what are they?
In medicine, this term refers to a large group of diseases transmitted at the genetic level from parent to child.
They are characterized by impaired muscle function, low motor activity or its absence.
Such consequences occur as a result of dysfunction of neuromuscular junctions, damage to muscles, spinal neurons or nerves.
The main causes of NMD are autoimmune diseases, poisoning with various substances and hereditary factors. Also included here are some congenital metabolic defects.
Classification
NMZ includes:
- primary progressive muscular dystrophies (myopathies);
- secondary progressive muscular dystrophies;
- congenital non-progressive myopathies;
- myotonia;
- hereditary paroxysmal myoplegia.
We'll talk about each type in more detail later.
Primary progressive muscular dystrophies (myopathies)
Signs of progressive muscular dystrophy (PMD) include degenerative changes in the muscles.
They consist in thinning the muscles, replacing muscle tissue with fatty and connective tissue.
Focal necrosis occurs, and transverse striations are lost.
Some mechanisms of the onset and course of the disease have not yet been established. Myopathy is caused by membrane defects in muscle cells. Great hopes for establishing the details of pathogenesis are placed on microbiologists.
With myopathies, the muscles weaken and then atrophy. There are several forms of this disease. Different types of PMD are distinguished by the type of inheritance, timing of manifestation, nature and course of the disease, and location of atrophies.
Hereditary paroxysmal myoplegia
Paroxysmal myoplegia is a rare hereditary disease in children. Characterized by periodic attacks of paralysis of the skeletal muscles. It is inherited according to the principle in which one mutant allele localized in the autosome is sufficient for the manifestation of the disease.
There are three forms:
- hyperkalemic;
- hypokalemic;
- normokalemic.
The hypokalemic form of paroxysmal myoplegia most often affects men.
The first attacks of the disease appear at the age of 10.
The attack occurs in the morning or at night. In this case, weakness is felt in the limbs and neck, often reaching paralysis. In some cases, paralysis spreads to the facial muscles and respiratory tract.
The attack is accompanied by thirst, hyperemia, and sweating. The duration can be from an hour to seven days. Women usually experience an attack on the first day of their period.
The hyperkalemic form can be found less frequently; its attacks begin at a young age. The attack is provoked by cold and prolonged muscle inactivity. It begins with a sensitivity disorder in the facial muscles, arms and legs.
The third form of the disease manifests itself before the age of 10. The attacks go away within a few days or weeks. They can be triggered by low ambient temperature, alcohol consumption, and heavy physical activity.
Reference! Hereditary paroxysmal myoplegia can be symptoms of diseases of the thyroid gland, Addisson, and are less common with vomiting and diarrhea.
Congenital non-progressive myopathies
Non-progressive myopathy is benign and appears in infancy or childhood. This type of NMD includes diseases of the central core, namaline and mitochondrial myopathies. In this case, metabolic muscle changes occur. The patient feels a decrease in strength and weakened reflexes.
Myotonia
Myotonia is a delayed relaxation of a muscle after tension.
The disease is divided into three types:
- action myotonia;
- percussion myotonia;
- electromyographic myotonia.
If a patient with action myotonia clenches his fingers into a fist and then tries to quickly straighten them, he will need time to fully straighten his palm.
The second type is characterized by muscle contraction during intense tapping with a hammer.
To detect electromyographic myotonia, a needle shock will be required. At the same time, the device shows high-frequency discharges, which are at first more frequent and then decrease in frequency.
List of all NMZ
- Diseases of the central nervous system;
- Thomsen's myotonia;
- Steinert-Kurschmann disease;
- Becker's muscular dystrophy;
- Duchenne muscular dystrophy;
- Landouzy-Dejerine myodystrophy;
- Mitochondrial myopathies;
- Neural amyotrophies of Charcot-Marie-Tooth;
- Nemaline myopathies;
- Ophthalmoplegic myopathies;
- Werdnig-Hoffmann spinal amyotrophy;
- Kugelberg-Welander spinal amyotrophy;
- Congenital muscular dystrophy;
- Becker muscular dystrophy;
- Pompe disease;
- Myotonic dystrophy;
- Duchenne muscular dystrophy.
Treatment options
Treatment is aimed at supporting muscle strength and slowing down atrophy.
The goal of therapy is for the patient to move around without assistance for as long as possible, because... In a constant vertical position, breathing disorders occur.
The main methods of treatment are:
- Medical and physical training complex. These are various active and passive movements. The patient should not be subjected to increased stress. Exercises should be regular;
- Orthopedic measures. The use of special tires and operations. These measures are aimed at preserving independent movement;
- Medications. Medicines that support metabolism, eliminating energy and protein deficiency. Patients are prescribed phosphaden, prednisolone, nifedipine, vitamin E.
An incorrect preliminary diagnosis leads to errors in diagnosing the form of NMD. This increases the cost of expensive tests and hinders the prevention of relapse, determination of the genetic status of the patient’s ancestors and the selection of adequate pathogenetic treatment, which can be limited to in some cases.
on this topic:
Source: //doktor-ok.com/zabolevaniya/nervno-myshechnye
Symptoms of femoral nerve neuropathy
The clinical symptom complex of femoral neuropathy depends on the topic of the process. When pathology occurs at the iliopsoas level, a full range of symptoms develops, including sensory, motor and autonomic-trophic disorders throughout the area innervated by the femoral nerve. In rare cases, with a high division of the nerve, only sensory or only motor disturbances can be observed, sometimes a mosaic picture of motor and sensory disturbances.
Complete neuropathy of the femoral nerve is accompanied by only partial disruption of the iliopsoas muscles, due to the existence of their alternative innervation. Therefore, flexion and supination of the hip are practically not impaired. Paresis of the quadriceps muscle, which is responsible for straightening the leg at the knee joint, is more pronounced. Due to difficulty in extension, patients try not to bend their leg at the knee. Running and walking are difficult, especially when it is necessary to climb stairs. The gait changes. The leg is fixed in a hyperextension position. There is a lack of knee reflex.
Sensory disorders include disorders of tactile and pain perception on the anterior inner surface of the thigh and lower leg, and the medial edge of the foot. In the same zone, trophic and vegetative changes are observed, and irritating pain is possible. When lying on the stomach, symptoms of tension are detected - pain along the front surface of the thigh when trying to raise a straight leg as much as possible (Wassermann's symptom) or bend the leg at the knee joint (Mickiewicz symptom).
Neuropathy of the femoral nerve when it is affected in the area of the inguinal ligament is in general similar to the clinical picture described above. With a high origin of the saphenous nerve, predominantly movement disorders can be observed. Along with the symptoms of tension, pain is detected when pressing in the middle of the inguinal ligament.
Compression of the femoral nerve trunk in Gunter's canal is characterized by painful and tactile hypoesthesia of the skin of the medial edge of the knee joint, the anterior inner surface of the leg and the inner edge of the foot. In the same area, paresthesia and pain are observed, which increase in intensity when the lower leg is extended. The latter forces the patient to walk and stand with the leg slightly bent at the knee. The knee reflex is not impaired. Pain is detected at the point of exit of the saphenous nerve from the adductor canal, Tinel's symptom is the appearance of paresthesia along the nerve when it is tapped with a neurological hammer.
Neuromuscular diseases
Neuromuscular diseases are a conditionally distinguished group of diseases that are characterized by impaired muscle function, primarily by their weakness.
Neuromuscular diseases include muscle diseases, peripheral nerve diseases, neuromuscular junction diseases and motor neuron diseases.
The same symptom of muscle weakness can be a manifestation of diseases with very different mechanisms. This determines a completely different prognosis and treatment methods.
Acquired myopathies:
- inflammatory myopathies: (polymyositis, dermatomyositis, inclusion body myositis, sarcoid myopathy;
- infectious myopathies (myopathies with HIV, viral myositis, bacterial myositis, parasitic myositis);
- drug and toxic myopathies (corticosteroid myopathy, myopathy when using drugs to lower cholesterol, alcoholic myopathy, myopathy in critical conditions).
- hypokalemic myopathy;
- hypophosphatemic myopathy;
- myopathy in chronic renal failure;
- myopathy in diabetes;
- myopathy in hypothyroidism;
- myopathy in hyperthyroidism;
- myopathy due to hyperparathyroidism;
- Cushing's disease.
- myoglobinuria;
- channelopathies;
- hereditary myopathies;
- muscular dystrophy.
Peripheral nerve diseases
Neuromuscular junction diseases
- Myasthenia gravis
- Lambert-Eaton syndrome
- Botulism
- Tick paralysis
Motor neuron diseases
- Amyotrophic lateral sclerosis
- Lower Motor Neuron Diseases
- spinal muscular atrophy
- monomelic amyotrophic lateral sclerosis
- Kennedy's disease
- Upper motor neuron diseases
- hereditary spastic paraparesis
- primary lateral sclerosis
The neuromuscular junction or neuromuscular synapse is the connection of a nerve ending and a muscle fiber to form the so-called synaptic cleft, in which impulses are transmitted from the nerve to the muscle membrane.
The impulse is transmitted using the neurotransmitter acetylcholine, released at the end of the nerve and then attached to the muscle membrane.
In some diseases, neuromuscular transmission is disrupted due to insufficient release of acetylcholine from the nerve ending or due to disruption of its attachment to the muscle fiber membrane.
Myasthenia Gravis
The Greek term myasthenia gravis translates as “muscle weakness” and gravis as “severe.” Myasthenia Gravis is a disease characterized by severe muscle weakness and fatigue.
With myasthenia gravis, there is a disruption in the transmission of impulses from the nerve fiber to the muscle fiber.
The disease is based on the production of autoantibodies that block the attachment of the neurotransmitter acetylcholine to the muscle membrane at the neuromuscular junction.
How is the diagnosis made?
The diagnosis of myasthenia gravis is made by a doctor based on a blood test and electroneuromyography. If necessary, a computed tomography scan of the chest is prescribed to assess the size and condition of the thymus gland as a search for a possible cause of the disease (production of autoantibodies).
Treatment
In the treatment of myasthenia gravis, anticholinesterase drugs (Pyridostigmine or Kalimin) and drugs that suppress the immune system (prednisolone and others) are used. Removal of the thymus gland (thymectomy) is performed when drug therapy is ineffective. Plasmapheresis and immunoglobulins can also be used in treatment.
Lambert-Eaton syndrome
Lambert-Eaton syndrome is a syndrome of muscle weakness and fatigue that develops due to an autoimmune process. Usually the cause of the syndrome is a malignant oncological process, most often lung cancer. Therefore, when making a diagnosis of Lambert-Eaton syndrome, the patient is always indicated for further examination for the purpose of cancer detection.
Why does Lambert-Eaton syndrome occur?
The cause is antibodies produced by the body itself (a similar autoimmune conflict is observed in myasthenia gravis). In particular, antibodies destroy nerve endings, thereby disrupting the regulation of the amount of neurotransmitter released.
When there is not enough neurotransmitter, the muscles cannot contract. The disease is not hereditary; it mainly affects young people under 40 years of age. The prevalence of the disease is 1 per 1,000,000 people.
40% of patients with Lambert-Eaton syndrome are diagnosed with cancer.
How is Lambert-Eaton syndrome diagnosed?
Diagnostics includes a blood test for antibodies, test administration of an anticholinesterase drug, and electroneuromyography.
Myasthenia gravis
The disease most often affects women (2/3 of the total number of patients). Myasthenia has two forms - congenital and acquired. With this disease, the transmission of nerve impulses is disrupted, resulting in weakness in the striated muscles.
The disease is associated with changes in the functions of the neuromuscular system. Weakening muscles affect the normal functioning of organs: the patient may have constantly half-closed eyelids, difficulty urinating, and difficulties in chewing and walking. As a result, the disease can lead to disability and even death.
Stages and degrees
Neuromuscular diseases occur in stages. A neurologist will help determine the stage of development of pathological processes using medical diagnostics.
| Name | Description |
| Stage I | Motor disturbances are mild. |
| Stage II | The patient has pronounced clinical signs and serious motor changes. |
| Stage III | The patient cannot move independently. |
Neuromuscular diseases Duchenne myopathy at stage 2.
The clinical picture depends on the speed of development of pathological processes and the severity of the disease. A neurologist will help you establish an accurate diagnosis.