История открытия
Британский офтальмолог Уорнер Тей и американский невролог Бернард Сакс независимо друг от друга описали заболевание в 1887 году и разработали критерии диагностики, позволяющие отличить данную болезнь от других неврологических нарушений с похожими симптомами.
Бернард Сакс первым выдвинул гипотезу о том, что данная патология является генетической. Его интуитивное предположение подтвердилось в середине ХХ века после переоткрытия законов Менделя.
Бернард Сакс предложил для нового заболевания название, которое можно встретить и в современной медицинской литературе — амавротическая семейная идиотия.
Библиография
Бадалян Л. О., Таболин В. А. и Вельтищев Ю. Е. Наследственные болезни у детей, с. 82, М., 1971; Гербер Э. Л. и Мошковская Р. Д. Поздняя форма амавротической идиотии, Сб. науч. трудов, посвящ. 150-летию Моск. психоневрол. б-цы № 3, в. 4, с. 365, 1963; Мнухин С. С. Амавротическая идиотия, Многотомн. руководство по неврол., под ред. С. Н. Давиденкова, т. 7, с. 366, Л., 1960; Bergener M. u. Jungklaass F. К. Beitrag zur Klinik und Genetik der amaurotischen Idiotie (Spatinfantile und Spatform) unter besonderer Beriicksichtigung diagnostischer und differentialdiagnostischer Probleme, Nervenarzt, S. 316, 1968; Cerebral sphingolipidoses, ed. by M. A. Stanley a. B. W. Volk, N. Y.—L., 1962, bibliogr.; Jatzkewitz H. Evidence for metabolic blocks in sphingolipidoses, Proc. 5-th int. congr. neuropathol., p. 417, Amsterdam a. o., 1966; Schettler G. Gangliosidosen, Handb. inn. Med., hrsg. v. G. Bergmann u. a., Bd 7, T. 2, S. 657, В. u. a., 1955, Bibliogr.; Schneck L. Volk B. W. a. Saifer A. The gangliosidoses, Amer. J. Med., v. 46, p. 245, 1969, bibliogr.; Stanbury J. В. а. о. The metabolic basis of inherited disease, N. Y. a. o., 1966; Suzuku J. Y. a. o. Gm2-gangliosidosis with total hexosaminidase deficiency, Neurology (Minneap.), v. 21, p. 313, 1971, bibliogr.
Л. О. Бадалян; В. В. Ковалев (психиат.).
Источник: Большая Медицинская Энциклопедия (БМЭ), под редакцией Петровского Б.В., 3-е издание
Причины заболевания
Долгое время врачи не могли дать ответ на вопрос, что же вызывает болезнь Тея-Сакса. Причины патологии стали известны только в середине ХХ века, когда сформировались представления о генетике. Исследования показали, что болезнь развивается в результате мутации гена НЕХА, который располагается на 15-ой хромосоме. Заболевание является разновидностью GM2-ганглиозидоза — генетической патологии, связанной с недостатком или пониженной активностью гексозаминидазы. Амавротическая идиотия возникает в результате снижения активности гексазаминидазы А или недостатка этого фермента.
Заболевание передается по аутосомно-рецессивному типу, потому если в генотипе человека есть здоровый гена НЕХА, то у него не проявляется болезнь Тея-Сакса. Генетика заболевания схожа с наследованием таких патологий как болезнь Гоше, болезнь Урбаха-Вите, синдром Дабина-Джонсона: если оба родителя являлись носителями мутировавшего гена, вероятность рождения больного ребенка 0,25%, если и мать, и отец были больны, то и у детей заболевание проявляется почти в 100% случаев.
Этиология и патогенез амавротической идиотии
В основе заболевания лежит энхимноз, нарушение обмена липидов, ведущее к отложению в нервной системе липида ганглиозида. У здоровых людей ганглиозид определяется только в сером веществе мозга. Строение ганглиозида сложное: он содержит жирные кислоты — преимущественно стеариновую, нейраминовую, сфингозин, а также сахарозу, галактозу, глюкозу.
Установлено, что при амавротической идиотии имеется глубокое нарушение липидного обмена. Наиболее полно изучена амавротическая идиотия Тея—Сакса, которую отнесли к группе внутриклеточных липоидозов. Кленк (Е. Klenk, 1939, 1942) отметил при болезни Тея—Сакса увеличение в мозге гликолипида-ганглиозида. Нормен (R. М. Norman, 1963), Шнек (L. Schneck, 1969), Судзуки (Y.
Suzuky, 1971) выявили увеличение содержания ганглиозидов в печени, селезенке, эритроцитах крови, что свидетельствовало о генерализованном нарушении обмена ганглиозидов. Нарушение метаболизма ганглиозида GM2 связывают с отсутствием или резким снижением содержания энзима гексозаминидазы-А (Шнек, 1970; Судзуки, 1971). Отсутствие этого фермента отмечается уже у эмбриона на 18—20-й неделе. У гетерозиготных носителей гена амавротической идиотии Тея—Сакса отмечается умеренное снижение гексозаминидазы-А.
Особенности нарушений обмена веществ при других формах амавротической идиотии и, в частности, является ли для них нарушение биохимических процессов первичным, точно еще не установлено. Предполагается, что при врожденной форме Нормена — Вуда имеет место отложение ганглиозида типа GM3 [Осетовская (Е. Osetowska, 1968)]. При ювенильной форме Баттена — Шпильмейера—Фогта, по мнению ряда авторов, накапливаются ганглиозиды в смеси с холестерином и липопигмент типа липофусцина.
Анатомической базой для развития заболевания считается специфический распад ганглионарных клеток, появляющийся как следствие нарушения липидного обмена внутри клетки.
Основные формы болезни
Принято выделять три основных формы заболевания. Наиболее распространенная из них — младенческая. Дети с болезнью Тея-Сакса развиваются нормально до 6-7 месяцев. После этого начинается медленный, но необратимый процесс снижения умственных и физических способностей.
Существует также ювенальная форма заболевания. По сравнению с младенческой, она встречается реже. Примерно до 3-10 лет ребенок развивается так же, как и его сверстники, но со временем начинается медленное снижение когнитивных и двигательных функций, развивается дизартрия, дисфагия, атаксия.
Болезнь Тея-Сакса с поздним началом — наиболее редкая форма заболевания. Первые признаки заболевания проявляются обычно после 30 лет. Однако зафиксированы случаи и более раннего появления симптомов (15-18 лет). Эта форма заболевания имеет самый благоприятный прогноз, так как ее прогрессирование можно остановить.
Симптомы
Независимо от формы заболевания, проявляется несколько основных симптомов: дисфагия, атаксия, утрата когнитивных функций, мышечная атрофия. Если ребенок до года остро реагирует на резкие звуки, плохо набирает вес и не может расслабить мышцы, родителям стоит показать его специалистам — именно так у младенцев начинается болезнь Тея-Сакса. Симптомы становятся все более серьезными. После 6 месяцев снижается двигательная активность, младенец теряет способность самостоятельно сидеть и менять позу. Постепенно развивается слепота, снижается слух, атрофируются мышцы и развивается полный паралич тела.
При ювенальной форме, помимо основных симптомов, наблюдается дизартрия (снижение четкости речи), спастичность, нарушение координации движений. Происходит постепенная утрата когнитивных функций — снижение памяти, внимания, работоспособнсти. Развивается деменция. На поздних стадиях болезни появляются судороги.
Первые симптомы взрослой формы заболевания — затрудненное глотание, нарушение координации и дизартрия. Часто появляются психические нарушения схожие с симптомами шизофрении (зрительные и слуховые галлюцинации, апатия, снижение эмоциональности). Без лечения наблюдается ухудшение когнитивных функций. Только для данной формы заболевания существует эффективное лечение, позволяющее остановить болезнь Тея-Сакса. Национальное руководство по неонатологии говорит о том, что эффективные методы диагностики взрослой формы заболевания появились только в 70-е, до этого заболевание считалось детским.
Клиническая картина
Врожденная форма Нормена—Вуда проявляется в первые дни или недели после рождения. У ребенка обнаруживаются гидроцефалия или микроцефалия, судороги, параличи, расстройства дыхания, глотания. Летальный исход обычно наступает в первые месяцы жизни.
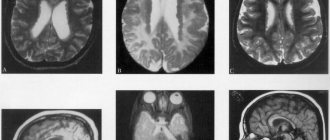
Ранняя детская (инфантильная) форма — болезнь Тея—Сакса — начинается в возрасте 4—6 месяцев. Часто заболевание носит семейный характер. Рано обнаруживается снижение зрения. Ребенок не может фиксировать взгляд, не следит за игрушками. Довольно рано на глазном дне обнаруживается симптом «вишневой косточки» — вишнево-красное пятнышко в макулярной области, окруженное серовато-белым ободком.
В последующем развивается атрофия зрительных нервов и полная слепота. Исчезают ориентировочные и защитные реакции. Двигательные нарушения приводят к полной обездвиженности. При болезни Тея—Сакса наблюдается симптом повышенной реакции на звуковые раздражения — дети резко вздрагивают от обычного звука, могут отмечаться судороги, носящие преимущественно тонический характер. В конечной стадии болезни развивается кахексия и состояние децеребрационной ригидности. Смерть наступает в среднем через 11/2—2 года после начала заболевания.
Поздняя детская форма Бильшовского—Янского начинается в 3—4 года. Течение прогрессирующее, с ремиссиями. Нарастающая органическая деменция сочетается с общими судорожными припадками, атаксией, экстрапирамидными расстройствами. На глазном дне выявляется атрофия зрительных нервов. Некоторые авторы отрицают нозологическую самостоятельность поздней детской формы Бильшовского—Янского и рассматривают случаи этого заболевания как проявление рано начинающейся ювенильной формы Баттена—Шпильмейера—Фогта пли как более поздние варианты болезни Тея—Сакса. Летальный исход в большинстве случаев наступает в конце первого десятилетия жизни.
Юношеская форма Баттена—Шпильмейера—Фогта начинается в возрасте 6—10 лет, характеризуется медленно прогрессирующим течением. Картина глазного дна часто соответствует пигментному ретиниту. Заболевание начинается с постепенного падения зрения и нарастающей деменции. Изменения в двигательной системе могут быть различными и непостоянными: наблюдаются нерезко выраженные тетрапарезы, экстрапирамидные и бульбарные нарушения. Нередок эпилептиформный синдром. Заболевание заканчивается смертельным исходом на 2—3-м десятилетии жизни.
Поздняя амавротическая идиотия Куфса наблюдается крайне редко, возникает в зрелом возрасте и характеризуется вялым течением. Развиваются изменения личности по типу органического психического синдрома. На глазном дне выявляется картина пигментного ретинита с атрофией зрительных нервов. В поздней стадии заболевания развиваются параличи и эпилептиформный синдром. Больные обычно погибают в течение 10—15 лет от начала заболевания.
Психические расстройства при разных формах амавротической идиотии имеют различия, отчасти связанные с возрастными особенностями.
При врожденной форме Нормена—Вуда в первые недели жизни наблюдается остановка нервно-психического развития.
При ранней детской форме Тея—Сакса нормально развивавшиеся до того дети относительно быстро становятся вялыми, утрачивают интерес к окружающему, перестают узнавать близких. Происходит постепенное исчезновение приобретенных двигательных навыков и речи. В течение нескольких месяцев, реже до 2 лет, развивается глубокое слабоумие типа идиотии.
При поздней детской форме Бильшовского—Янского психическая деградация происходит более медленно, чем при ранней детской форме. Может наблюдаться волнообразное течение с периодами ухудшения (в виде вялости, апатии, оглушения) и улучшения (с частичным восстановлением активности). В конечной стадии имеет место глубокое слабоумие с утратой речи и навыков самообслуживания.
При юношеской форме Баттена—Шпильмейера—Фогта наблюдаются медленно нарастающие вялость, апатия, обеднение речи. Все более заметно интеллектуальное снижение, однако редко достигающее степени идиотии, нарушения памяти. Присоединяется дизартрия, нарушаются навыки письма и чтения. В конечной стадии наблюдается выраженная органическая деменция.
При поздней форме Куфса психические расстройства отличаются наибольшей вариабельностью. Для нее характерен наиболее медленный темп прогрессирования. Начальные проявления выражаются изменениями личности в виде снижения активности, сужения круга интересов. Спустя несколько лет все более заметным становится прогрессирующее снижение интеллекта вплоть до органической деменции с апатией или легкой эйфорией, потеря навыков самообслуживания. Описываются случаи с галлюцинаторно-бредовыми и кататоническими расстройствами, а также форма с относительно сохранным интеллектом.
Постановка диагноза
Врачам не всегда удается поставить верный диагноз, если речь идет о такой редкой патологии как болезнь Тея-Сакса. Симптомы, генетика и лечение заболевания активно изучаются специалистами. Независимо от формы болезни, существует несколько диагностических процедур, которые проводятся при подозрении на ее наличие. Одна из них — определение активности фермента гексозаминидазы в сыворотке крови, лейкоцитах или фибробластах. У пациентов с болезнью Тея-Сакса активность гексозаминидазы В всегда ниже нормы, фермент гексозаминидаза А практически отсутствует или его активность значительно ниже нормальной.
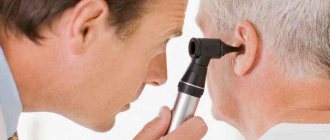
Еще один важный диагностический критерий — наличие ярко-красного пятна на роговице глаза, которое при помощи офтальмоскопа легко заметит терапевт или окулист. Красное пятно на роговице было обнаружено у всех пациентов, независимо от возраста.
В отличие от других лизосомных болезней накопления (болезнь Гоше, синдром Стандхоффа, болезнь Ниманна-Пика), при болезни Тея-Сакса не происходит увеличение печени и селезенки (гепатоспленомегалия).
Лечение
На данный момент не существует препаратов, позволяющих вылечить болезнь Тея-Сакса. Симптомы и лечение заболевания до сих пор являются предметом научных исследований.
Младенческая форма болезни Тея-Сакса наиболее опасна. Если больной ребенок не может самостоятельно глотать, рекомендуется прибегают к искусственному питанию, восстановить физические навыки невозможно. Никаких препаратов, способных остановить или повернуть вспять развитие заболевания не существует, несмотря на все старания ученых. Больные младенцы, даже если они получают наилучший уход, редко доживают до четырехлетнего возраста.
При ювенальной форме заболевания важно, чтобы ребенок постоянно находится под наблюдением врача. Следование указанием специалиста и прохождение всех необходимых медицинских процедур помогает продлить жизнь больного ребенка до 12-16 лет.
Взрослая форма заболевания прогрессирует медленнее других и часто поддается лечению. При психических нарушениях пациентам назначают препараты лития или хлорид цезия. Клинические испытания показали, что пириметамин позволяет существенно замедлить, а в редких случаях и полностью остановить прогрессирование болезни за счет увеличения активности гексозаминидазы В.
Осложнения
На начальных этапах заболевания двигательные и зрительные нарушения обуславливают частую травматизацию ребёнка. Резкое уменьшение двигательной активности провоцирует развитие застойной пневмонии. Осложнением судорожного синдрома является эпилептический статус. Дисфагия при бульбарном синдроме опасна попаданием жидкости и пищи в дыхательные пути с возникновением аспирационной пневмонии.
https://www.youtube.com/watch?v=watch
Целый ряд осложнений связан с отложением липидов во внутренних органах. Жировая дистрофия печени ведёт к развитию печёночной недостаточности, жировые отложения на створках клапанов — к сердечной недостаточности. Пациенты погибают от сердечной, дыхательной, полиорганной недостаточности, интеркуррентных инфекций.
В число симптомов БТС входят эпилептические приступы, представляющие собой результат внезапных вспышек аномальной биоэлектрической активности мозга. При их высокой частоте физическая и психическая деградация происходит быстрее. Во время приступа больные падают, бьются в конвульсиях, что сопровождается высоким риском удушья (западение корня языка), получения смертельных травм.
Пренатальная диагностика
Современные исследования еще на раннем строке беременности позволяют определить, унаследовал ли ребенок мутантный ген НЕХА от родителей. Если оба родителя являются носителями заболевания, рекомендуется произвести биопсию хориона. Это одна из самых распространенных процедур пренатальной диагностики, цель которой — выявить генетические аномалии плода. Проводится на 10-14 неделе беременности. Амниоцентез также дает четкое представление о том, является и ребенок носителем мутировавшего гена НЕХА. Данные процедуры имеют риск выкидыша менее 1%.
В случае искусственного оплодотворения можно определить генетические аномалии плода еще до его имплантации в матку. Для этого проводится преимплантационная генетическая диагностика, аналог пренатальной диагностики. Ее основной плюс в том, что процедура не инвазивная и абсолютно безопасная. Можно отобрать только здоровые эмбрионы для имплантации, тем самым снизив практически до нуля риск рождения ребенка с болезнью Тея-Сакса.