History of discovery
British ophthalmologist Warner Tay and American neurologist Bernard Sachs independently described the disease in 1887 and developed diagnostic criteria to distinguish the disease from other neurological disorders with similar symptoms.
Bernard Sachs was the first to hypothesize that this pathology is genetic. His intuitive assumption was confirmed in the mid-twentieth century after the rediscovery of Mendel's laws.
Bernard Sachs proposed a name for the new disease that can also be found in modern medical literature - amaurotic familial idiocy.
Bibliography
Badalyan L. O., Tabolin V. A. and Veltishchev Yu. E. Hereditary diseases in children, p. 82, M., 1971; Gerber E. L. and Moshkovskaya R. D. Late form of amaurotic idiocy, Sat. scientific works, dedicated 150th anniversary of Moscow. psychoneurol. b-tsy No. 3, v. 4, p. 365, 1963; Mnukhin S.S. Amaurotic idiocy, Multivolume. Guide to neurol., ed. S. N. Davidenkova, vol. 7, p. 366, L., 1960; Bergener M.u. Jungklaass F. K. Beitrag zur Klinik und Genetik der amaurotischen Idiotie (Spatinfantile und Spatform) unter besonderer Beriicksichtigung diagnostischer und differentialdiagnostischer Probleme, Nervenarzt, S. 316, 1968; Cerebral sphingolipidoses, ed. by MA Stanley a. BW Volk, NY-L., 1962, bibliogr.; Jatzkewitz H. Evidence for metabolic blocks in sphingolipidoses, Proc. 5th int. congr. neuropathol., p. 417, Amsterdam ao, 1966; Schettler G. Gangliosidosen, Handb. inn. Med., hrsg. v. G. Bergmann ua, Bd 7, T. 2, S. 657, V. ua, 1955, Bibliogr.; Schneck L. Volk BW a. Saifer A. The gangliosidoses, Amer. J. Med., v. 46, p. 245, 1969, bibliogr.; Stanbury J. V. a. O. The metabolic basis of inherited disease, NY ao, 1966; Suzuku JY ao Gm2-gangliosidosis with total hexosaminidase deficiency, Neurology (Minneap.), v. 21, p. 313, 1971, bibliogr.
L. O. Badalyan; V. V. Kovalev (psychiatrist).
Source: Great Medical Encyclopedia (BME), edited by Petrovsky B.V., 3rd edition
Causes of the disease
For a long time, doctors could not answer the question of what causes Tay-Sachs disease. The causes of the pathology became known only in the middle of the twentieth century, when ideas about genetics were formed. Studies have shown that the disease develops as a result of a mutation in the HEXA gene, which is located on the 15th chromosome. The disease is a type of GM2 gangliosidosis, a genetic pathology associated with a deficiency or decreased activity of hexosaminidase. Amaurotic idiocy occurs as a result of a decrease in the activity of hexazaminidase A or a deficiency of this enzyme.
The disease is transmitted in an autosomal recessive manner, therefore if a person has a healthy HEXA gene in his genotype, then he does not develop Tay-Sachs disease. The genetics of the disease is similar to the inheritance of such pathologies as Gaucher disease, Urbach-Wiethe disease, Dabin-Johnson syndrome: if both parents were carriers of the mutated gene, the probability of having a sick child is 0.25%, if both mother and father were sick, then both In children, the disease manifests itself in almost 100% of cases.
Etiology and pathogenesis of amaurotic idiocy
The disease is based on enchymnosis, a disorder of lipid metabolism leading to the deposition of ganglioside lipid in the nervous system. In healthy people, the ganglioside is detected only in the gray matter of the brain. The structure of the ganglioside is complex: it contains fatty acids - mainly stearic, neuraminic, sphingosine, as well as sucrose, galactose, and glucose.
It has been established that in amaurotic idiocy there is a profound disturbance of lipid metabolism. The most fully studied amaurotic idiocy is Tay-Sachs, which is classified as a group of intracellular lipoidoses. Klenk (E. Klenk, 1939, 1942) noted an increase in glycolipid ganglioside in the brain in Tay-Sachs disease. Normen (R. M. Norman, 1963), Schneck (L. Schneck, 1969), Suzuki (Y.
Suzuky, 1971) revealed an increase in the content of gangliosides in the liver, spleen, and red blood cells, which indicated a generalized disorder of ganglioside metabolism. Impaired metabolism of ganglioside GM2 is associated with the absence or sharp decrease in the content of the enzyme hexosaminidase-A (Schneck, 1970; Suzuki, 1971). The absence of this enzyme is noted already in the embryo at 18-20 weeks. Heterozygous carriers of the Tay-Sachs amaurotic idiocy gene exhibit a moderate decrease in hexosaminidase-A.
The features of metabolic disorders in other forms of amaurotic idiocy and, in particular, whether the disorder of biochemical processes is primary for them has not yet been precisely established. It is assumed that in the congenital form of Norman-Wood, deposition of a ganglioside of the GM3 type occurs [Osetovskaya (E. Osetowska, 1968)]. In the juvenile form of Batten-Spielmeyer-Vogt, according to some authors, gangliosides mixed with cholesterol and lipopigment such as lipofuscin accumulate.
The anatomical basis for the development of the disease is considered to be a specific breakdown of ganglion cells, which appears as a consequence of a violation of lipid metabolism within the cell.
Main forms of the disease
It is customary to distinguish three main forms of the disease. The most common of them is infantile. Children with Tay-Sachs disease develop normally until 6-7 months. After this, a slow but irreversible process of decline in mental and physical abilities begins.
There is also a juvenile form of the disease. Compared to infants, it is less common. Until approximately 3-10 years of age, the child develops in the same way as his peers, but over time, a slow decline in cognitive and motor functions begins, dysarthria, dysphagia, and ataxia develop.
Late-onset Tay-Sachs disease is the rarest form of the disease. The first signs of the disease usually appear after 30 years. However, cases of earlier onset of symptoms (15-18 years) have also been recorded. This form of the disease has the most favorable prognosis, since its progression can be stopped.
Symptoms
Regardless of the form of the disease, several main symptoms appear: dysphagia, ataxia, loss of cognitive function, muscle atrophy. If a child under one year of age reacts sharply to sharp sounds, gains weight poorly and cannot relax his muscles, parents should show him to specialists - this is how Tay-Sachs disease begins in infants. Symptoms become increasingly severe. After 6 months, motor activity decreases, the baby loses the ability to sit independently and change position. Blindness gradually develops, hearing decreases, muscles atrophy and complete paralysis of the body develops.
In the juvenile form, in addition to the main symptoms, dysarthria (decreased speech clarity), spasticity, and impaired coordination of movements are observed. There is a gradual loss of cognitive functions - a decrease in memory, attention, and performance. Dementia develops. In the later stages of the disease, seizures appear.
The first symptoms of the adult form of the disease are difficulty swallowing, loss of coordination and dysarthria. Mental disorders similar to the symptoms of schizophrenia (visual and auditory hallucinations, apathy, decreased emotionality) often appear. Without treatment, cognitive function deteriorates. Only for this form of the disease there is an effective treatment that can stop Tay-Sachs disease. The National Guide to Neonatology says that effective methods for diagnosing the adult form of the disease appeared only in the 70s; before that, the disease was considered a childhood disease.
Clinical picture
The congenital form of Norman-Wood appears in the first days or weeks after birth. The child exhibits hydrocephalus or microcephaly, convulsions, paralysis, breathing and swallowing disorders. Death usually occurs in the first months of life.
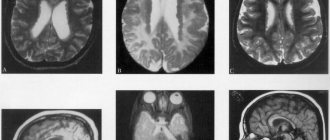
The early childhood (infantile) form - Tay-Sachs disease - begins at the age of 4-6 months. Often the disease is familial. Decreased vision is detected early. The child cannot fix his gaze and does not follow the toys. Quite early, the “cherry pit” symptom is detected in the fundus - a cherry-red spot in the macular area, surrounded by a grayish-white rim.
Subsequently, optic nerve atrophy and complete blindness develop. Indicative and defensive reactions disappear. Movement disorders lead to complete immobility. With Tay-Sachs disease, a symptom of an increased reaction to sound stimulation is observed - children flinch sharply from a normal sound, and convulsions may be observed, which are predominantly tonic in nature. In the final stage of the disease, cachexia and a state of decerebrate rigidity develop. Death occurs on average 11/2-2 years after the onset of the disease.
The late childhood form of Bielschowsky-Jansky begins at 3-4 years. The course is progressive, with remissions. Increasing organic dementia is combined with general convulsive seizures, ataxia, and extrapyramidal disorders. Atrophy of the optic nerves is detected in the fundus. Some authors deny the nosological independence of the late childhood form of Bielschowsky-Jansky and consider cases of this disease as a manifestation of the early onset juvenile form of Batten-Spielmeyer-Vogt or as later variants of Tay-Sachs disease. Death in most cases occurs at the end of the first decade of life.
The juvenile form of Batten-Spielmeyer-Vogt begins at the age of 6-10 years and is characterized by a slowly progressive course. The fundus picture often corresponds to retinitis pigmentosa. The disease begins with a gradual decline in vision and increasing dementia. Changes in the motor system can be different and inconsistent: mildly expressed tetraparesis, extrapyramidal and bulbar disorders are observed. Epileptiform syndrome is not uncommon. The disease ends in death in the 2nd—3rd decade of life.
Late amaurotic idiocy of Kufs is extremely rare, occurs in adulthood and is characterized by a sluggish course. Personality changes develop according to the type of organic mental syndrome. The fundus reveals a picture of retinitis pigmentosa with atrophy of the optic nerves. In the late stage of the disease, paralysis and epileptiform syndrome develop. Patients usually die within 10-15 years from the onset of the disease.
Mental disorders in different forms of amaurotic idiocy have differences, partly related to age characteristics.
In the congenital Norman-Wood form, neuropsychic development stops in the first weeks of life.
In early childhood Tay-Sachs form, previously normally developing children relatively quickly become lethargic, lose interest in their surroundings, and stop recognizing loved ones. There is a gradual disappearance of acquired motor skills and speech. Within a few months, rarely up to 2 years, profound dementia such as idiocy develops.
In the late childhood form of Bielschowsky-Jansky, mental degradation occurs more slowly than in the early childhood form. A wave-like course may be observed with periods of deterioration (in the form of lethargy, apathy, stupor) and improvement (with partial restoration of activity). In the final stage, profound dementia occurs with loss of speech and self-care skills.
In the juvenile form of Batten-Spielmeyer-Vogt, slowly increasing lethargy, apathy, and impoverished speech are observed. Intellectual decline is becoming more and more noticeable, but rarely reaching the level of idiocy and memory impairment. Dysarthria develops, writing and reading skills are impaired. In the final stage, severe organic dementia is observed.
In the late form of Kufs, mental disorders are characterized by the greatest variability. It is characterized by the slowest rate of progression. Initial manifestations are expressed by personality changes in the form of decreased activity, narrowing the range of interests. After a few years, a progressive decline in intelligence up to organic dementia with apathy or mild euphoria, and loss of self-care skills become increasingly noticeable. Cases with hallucinatory-delusional and catatonic disorders are described, as well as a form with relatively preserved intelligence.
Establishing diagnosis
Doctors are not always able to make the correct diagnosis when it comes to such a rare pathology as Tay-Sachs disease. The symptoms, genetics and treatment of the disease are being actively studied by specialists. Regardless of the form of the disease, there are several diagnostic procedures that are performed if its presence is suspected. One of them is the determination of the activity of the enzyme hexosaminidase in blood serum, leukocytes or fibroblasts. In patients with Tay-Sachs disease, the activity of hexosaminidase B is always below normal, the enzyme hexosaminidase A is practically absent or its activity is significantly below normal.
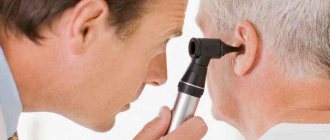
Another important diagnostic criterion is the presence of a bright red spot on the cornea of the eye, which can be easily noticed by a therapist or ophthalmologist using an ophthalmoscope. A red spot on the cornea was found in all patients, regardless of age.
Unlike other lysosomal storage diseases (Gaucher disease, Standhoff syndrome, Niemann-Pick disease), Tay-Sachs disease does not cause enlargement of the liver and spleen (hepatosplenomegaly).
Treatment
There are currently no drugs to cure Tay-Sachs disease. The symptoms and treatment of the disease are still the subject of scientific research.
The infant form of Tay-Sachs disease is the most dangerous. If a sick child cannot swallow on his own, it is recommended to resort to artificial nutrition; it is impossible to restore physical skills. There are no drugs that can stop or reverse the development of the disease, despite all the efforts of scientists. Sick babies, even if they receive the best care, rarely survive past the age of four.
In the juvenile form of the disease, it is important that the child is constantly under medical supervision. Following the instructions of a specialist and undergoing all necessary medical procedures helps to extend the life of a sick child to 12-16 years.
The adult form of the disease progresses more slowly than others and is often treatable. For mental disorders, patients are prescribed lithium or cesium chloride. Clinical trials have shown that pyrimethamine can significantly slow down, and in rare cases completely stop, the progression of the disease by increasing the activity of hexosaminidase B.
Complications
At the initial stages of the disease, motor and visual disturbances cause frequent trauma to the child. A sharp decrease in physical activity provokes the development of congestive pneumonia. A complication of convulsive syndrome is status epilepticus. Dysphagia with bulbar syndrome is dangerous due to the entry of liquid and food into the respiratory tract with the occurrence of aspiration pneumonia.
https://www.youtube.com/watch?v=watch
A number of complications are associated with lipid deposition in internal organs. Fatty liver leads to the development of liver failure, fatty deposits on the valve flaps lead to heart failure. Patients die from cardiac, respiratory, multiple organ failure, and intercurrent infections.
Symptoms of BTS include epileptic seizures, which are the result of sudden bursts of abnormal bioelectrical activity in the brain. With their high frequency, physical and mental degradation occurs faster. During an attack, patients fall and convulse, which is accompanied by a high risk of suffocation (retraction of the tongue root) and fatal injuries.
Prenatal diagnosis
Modern research makes it possible to determine early in pregnancy whether the child has inherited the mutant HEXA gene from its parents. If both parents are carriers of the disease, it is recommended to perform a chorionic villus biopsy. This is one of the most common prenatal diagnostic procedures, the purpose of which is to identify genetic abnormalities in the fetus. It is carried out at 10-14 weeks of pregnancy. Amniocentesis also gives a clear idea of whether the child is a carrier of the mutated HEXA gene. These procedures have a risk of miscarriage of less than 1%.
In the case of artificial insemination, genetic abnormalities of the fetus can be detected even before it is implanted in the uterus. For this purpose, preimplantation genetic diagnosis is performed, an analogue of prenatal diagnosis. Its main advantage is that the procedure is non-invasive and absolutely safe. It is possible to select only healthy embryos for implantation, thereby reducing the risk of having a child with Tay-Sachs disease to almost zero.