What is Huntington's chorea
Huntington's chorea (Huntington's disease, Huntington's syndrome, Huntington's disease) is an inherited neurodegenerative disease. The word "trochea" comes from Greek and means dance. Outwardly it manifests itself as involuntary, chaotic movements of the arms, legs, torso and facial muscles, often reminiscent of a strange dance. The degree of their severity depends on the stage of the disease. In addition, a person with Huntington's disease may develop anxiety, irritability, and memory loss, which affects communication and interactions with others.
As Huntington's disease progresses, more nerve cells die in the brain.
It should be remembered that with Huntington's disease, patients often cannot control their emotions and behavior.
But external symptoms are not the only manifestation of the disease. As the disease progresses, the nerve cells responsible for various functions in the brain gradually die, which leads to a slow decrease in the volume of this organ.
Definition of disease
Huntington's chorea is a chronic disease of the nervous system that progresses slowly and is inherited.
The disease is characterized by mental disorders, increasing choreic hyperkinesis (unregulated involuntary movements that limit the patient’s motor activity), as well as dementia (dementia).
Chorea is a disorder of motor function, which manifests itself in the form of erratic, jerky movements beyond the control of the patient. These can be either hand gestures or facial gestures, grotesque in their manifestation.
The disease was first described in 1872 by the American psychiatrist George Huntington, after whom it was named.
What is the cause of the disease
The body of any person consists of cells, each of which has a nucleus. It contains 23 pairs of chromosomes, consisting of tightly packed DNA, which contains a large number of genes. The cause of the development of Huntington's disease is a mutation, that is, a violation of the structure of a gene located on the fourth chromosome and encoding the formation of a protein called huntingtin. Huntingtin is found in various organs, but it is most abundant in the brain. When a disease occurs in a gene, the number of copies of its section, consisting of the letters of the genetic code CAG, indicating the corresponding chemical substances, increases many times:
- cytosine;
- adenine;
- guanine
The cause of Huntington's disease is a mutation in the gene region coded CAG
Normally, these copies should be no more than 35, but in Huntington's disease their number is 36 or more. Thus, excess huntingtin begins to have a toxic effect on the body. The more CAG repeats, the earlier the symptoms of the disease appear.
Huntington's chorea is considered a rare disease. As a rule, there are less than 10 cases per 100 thousand people.
Huntington's disease is inherited from a mutation carrier with a 50% probability. Both men and women can get sick equally. When transmitted through the paternal line, the severity of the mutation, that is, the number of CAG repeats, may increase, which means that the disease will manifest itself in children at an earlier age compared to the father. If the mutation is not passed on to the next generation and all its representatives are not its carriers, then the inheritance of the disease stops.
Video: Huntington's disease
Genetic research
Genetic prerequisites are checked using a blood test. DNA determines the structure of molecules that control the level of protein compounds. Based on this indicator, the number of repetitions that have a negative effect on protein coagulation is determined. If the number of repetitions exceeds the norm, the likelihood of chorea occurring is checked. Symptoms of the disorder may not yet appear for many years, but genetic testing will already show a predisposition to the disease.
Differential diagnosis
Symptoms in adults and children
Huntington's disease typically causes motor, cognitive and psychiatric disorders with a wide range of signs and symptoms. Initial manifestations vary greatly among affected individuals. During the course of the disease, some disorders dominate or have a greater impact on functional abilities.
One of the most striking manifestations of Huntington's disease is movement disorders. Most often, chorea is observed with this disease - involuntary, erratic movements of the body, reminiscent of dancing. Other symptoms include:
- unintentional jerks;
A wriggling movement that resembles a strange dance is the main symptom of Huntington's disease.
- muscle stiffness or contracture (dystonia);
- slow or abnormal eye movements;
Involuntary, chaotic movements of the muscles of the face and eyes are one of the signs of the disease - disturbances in gait, posture and balance;
- difficulty physically speaking or swallowing;
- problems with independent movement.
Physical activity has a major impact on the ability of a person with Huntington's chorea to work, perform daily activities, communicate, and remain independent.
However, when the disease begins before the age of 20 (juvenile form), patients, on the contrary, experience lethargy and stiffness in the body. In addition, as it progresses, difficulties in maintaining balance, impaired speech clarity and difficulty swallowing food are added. The latter is one of the reasons for uncontrolled weight loss.
In addition to movement disorders, changes in behavior and mood can often develop, such as:
- anxiety;
- irritability;
- fatigue and insomnia;
- depression;
- suicidal behavior;
- social withdrawal;
- lack of interest in the outside world, etc.
Depression is not an accidental symptom; it is a natural consequence of impaired brain function due to pathological changes in it.
Memory and other cognitive components may decrease, a person experiences:
- difficulty organizing yourself, setting priorities, or focusing on tasks;
- lack of flexibility in thinking, behavior or action;
- Poor self-control, which can lead to violent outbursts, acting without thinking, and sexual promiscuity;
- inability to recognize one's own behavior;
- slowness of thinking and “searching” for words;
- problems in learning new information.
Patients often experience obsessive-compulsive disorders - obsessive thoughts and repetitive behavior. There is a mental disorder called mania, which causes a sharp increase in mood, hyperactivity, impulsive behavior and high self-esteem.
Manifestations of Huntington's disease in humans can lead to conflicts between the patient and people around him if they are not aware of the peculiarities of his health condition.
Not all of these symptoms will necessarily occur in every patient. Combinations of manifestations of Huntington's disease are very individual.
Features of the symptoms of the juvenile form of the disease
The onset and progression of Huntington's disease at a young age has its own characteristics. The early stage of chorea in adolescents is characterized by the following behavioral changes:
- decreased academic performance;
- loss of previously acquired knowledge and skills;
- problematic behavior.
The greater the number of CAG repeats the analysis shows, the sooner symptoms of the disease will appear in a person.
Physical changes are presented:
- gait disturbance due to muscle stiffness;
- fine motor disorder (handwriting changes);
- tremors or small involuntary movements;
- convulsive seizures.
If the described changes in motor function, emotional or cognitive disturbances occur, you should contact a neurologist for examination and diagnosis.
Diagnostics
Preliminary diagnosis is based on a physical examination and analysis of mental and neurological status. During the appointment, the neurologist performs simple tests to evaluate:
- reflexes;
- muscle strength and tone;
- coordination and balance abilities.
The doctor also checks:
- hearing;
- vision and motor activity of the eyeballs;
- touch.
The neurologist pays attention to the patient’s mental state, mood, and offers standardized assessment tests:
- memory;
- argumentation;
- intelligence;
- language function;
- orientation in space.
Additionally, a consultation with a psychiatrist will be required to obtain conclusions regarding:
- behavioral pattern;
- emotional background;
- qualities of thinking and judgment;
- ability to grasp information;
- functional state of the brain.
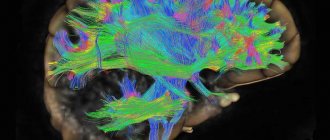
The main method of confirming the diagnosis of Huntington's disease is genetic testing to determine the number of CAG repeats. Additional examinations, such as magnetic resonance imaging (MRI) or computed tomography (CT), are used as needed if they influence patient management. MRI and CT scans can detect structural changes in certain areas of the brain affected by Huntington's disease, but only in later stages. These tests are used to rule out other conditions that may cause similar symptoms (eg, various dementias).
MRI reveals structural changes in areas of the brain affected by Huntington's disease
Genetic testing is also possible at the stage of carriage of the Huntington's disease mutation in the absence of external manifestations of the disease. Each person must make a voluntary, informed decision to carry it out based on the information received from the doctor dealing with the disease and in accordance with his own views in life.
Presymptomatic genetic testing for mutation carriers for Huntington's disease is not recommended for minors. To date, there are no methods for preventing or curing this disease, and the test results can have an extremely negative impact on the fate of the child, including the attitude of his family towards him, his ability to receive a proper education or choice of profession. Presymptomatic genetic testing should be delayed until adulthood, when the person can make an informed decision.
Features of nutrition and lifestyle
As Huntington's disease progresses, a person's functional abilities gradually deteriorate and they become increasingly dependent on caregivers. Eventually the patient will need constant assistance with eating and drinking. A person must be prepared for this in advance.
Patients with advanced Huntington's disease require assistance with daily tasks
There are other features that patients will encounter. In particular, issues related to nutrition and getting enough nutrients. People with Huntington's disease often have difficulty maintaining a healthy body weight. Trouble swallowing food, higher calorie needs due to exercise, or unknown metabolic problems can cause you to be thin.
To obtain adequate amounts of nutrients, meals should be taken 4-5 times a day with the use of dietary supplements. Problems can be minimized by removing distractions during meals and choosing foods that are easier to consume and digest.
Management of cognitive and mental disorders
Family and caregivers should try to create a comfortable environment for the person with Huntington's disease. Methods for structuring the patient's life include:
- using calendars and schedules to maintain a daily routine;
- initiating tasks using reminders;
- organization of any type of activity;
- overcoming tasks by breaking them down into small steps;
- creating a calm and friendly atmosphere;
- eliminating stressors that may cause outbursts of aggression, irritability, depression or other problems.
For school-aged children or adolescents, parental consultation with school personnel is necessary to develop an appropriate individualized education plan. Allowing the individual to maintain social interactions and friendships as much as possible is important.
Prognosis and complications
The rate of progression of the disease and its duration vary. The period from the onset of symptoms of Huntington's chorea to death ranges from 10 to 30 years. In the juvenile form, the disease usually leads to death within 10 years of the onset of symptoms.
Illness-related clinical depression may increase the risk of suicide. Studies show that the peak of a patient's desire to commit suicide occurs during the period of diagnosis and the middle stages of the disease, when a person begins to lose independence.
At the end of the illness, the patient is usually bedridden and unable to speak. However, people in this state are able to understand the language and address of family members and friends.
Common causes of death include:
- pneumonia or other infections;
- injuries associated with falls;
- complications associated with the inability to swallow.
Neurochemical changes
During a neurochemical study of the brain in patients suffering from Huntington's chorea, it was found that the concentration of GABA (aminobutyric acid, which is responsible for neurotransmitter and metabolic processes in the brain) in the striatum is significantly reduced.
The patients showed a decrease in choline acetyltransferase activity in the striatum, which may indicate a loss of cholinergic neurons. Although they are mostly preserved. The same applies to substance P, which is located in the middle styloid neurons of the striatum.
At the same time, the level of glutamate during chorea increases, as does the synthesis of cholinic acid.
You can find out what happens to the body with amyotrophic lateral sclerosis from our material. Taking the drug Carbamazepine is possible only when the instructions for use of the drug and reviews have been studied in detail.
You can find out what happens to the body with amyotrophic lateral sclerosis from our material. Taking the drug Carbamazepine is possible only when the instructions for use of the drug and reviews have been studied in detail.
Prevention
Individuals with a family history of Huntington's disease are understandably concerned about whether they may pass the mutation gene to their children. These individuals may consider genetic testing and family planning options. Today, there are approaches to family planning for Huntington's disease, thanks to which a carrier of a mutation of this disease can have a child without it.
If the mutation of the Huntington's disease gene is not passed on to the next generation, then inheritance of the disease stops
A parent with a gene mutation may benefit from meeting with a genetic counselor. The doctor will discuss the potential risks of a positive test result and talk about ways to keep future offspring safe, such as:
- prenatal gene testing;
- in vitro fertilization with donor sperm or egg.
Another option for couples is so-called in vitro fertilization and preimplantation genetic diagnosis. In this case, the egg is removed from the ovaries and fertilized with the father's sperm in the laboratory. Embryos are tested for the presence of the gene mutation, and only embryos that test negative are implanted in the mother's uterus.
Therapy
Huntington's chorea disease currently has no cure. Only symptomatic treatment has been developed, which helps improve the patient’s general condition and prolong his life. The following medications are used:
- Tetrabenazine was created specifically to reduce the severity of symptoms. However, this medicine has a side effect such as depression, so only a specialist can prescribe it.
- Neuroleptics alimemazine, periciazine, pipothiazine, thioproperazine and some others. These drugs help reduce the manifestations of pathology.
- Amantadine and remacemide are drugs that are still used as research drugs, but they have already shown good results.
- If seizures are present, valproic acid is used.
There is no surgical treatment for Huntington's chorea. More effective therapy is currently under development.
The prognosis is always unfavorable. From the moment the first symptoms of the pathology appear until death, 5 to 15 years pass. Moreover, death most often occurs not from this disease, but from its complications, among which pneumonia, heart problems and injuries are the leading ones. Also, according to statistics, more than half of patients with this disease die from suicide.