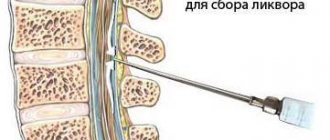
Brain development abnormalities
- the result of disturbances in the formation of individual cerebral structures or the brain as a whole that occur in the prenatal period. They often have nonspecific clinical symptoms: predominantly epileptic syndrome, mental and mental retardation. The severity of the clinical picture directly correlates with the degree of brain damage. They are diagnosed antenatally during obstetric ultrasound, after birth - using EEG, neurosonography and MRI of the brain. Treatment is symptomatic: antiepileptic, dehydration, metabolic, psychocorrective.
Brain development abnormalities
Anomalies of brain development are defects consisting of abnormal changes in the anatomical structure of cerebral structures. The severity of neurological symptoms accompanying cerebral anomalies varies significantly. In severe cases, defects are the cause of antenatal fetal death; they account for up to 75% of cases of intrauterine death. In addition, severe cerebral anomalies account for about 40% of newborn deaths. The timing of manifestation of clinical symptoms may vary. In most cases, cerebral abnormalities appear in the first months after the birth of the child. But, since the formation of the brain lasts until the age of 8, a number of defects make their clinical debut after the 1st year of life. In more than half of the cases, cerebral defects are combined with defects of somatic organs: congenital heart defects, renal fusion, polycystic kidney disease, esophageal atresia, etc. Prenatal detection of cerebral anomalies is an urgent task of practical gynecology and obstetrics, and their postnatal diagnosis and treatment are priority issues of modern neurology, neonatology, pediatrics and neurosurgery.
Brain Formation
The construction of the fetal nervous system begins literally from the first week of pregnancy. Already by the 23rd day of gestation, the formation of the neural tube ends, incomplete fusion of the anterior end of which entails serious cerebral anomalies. By approximately the 28th day of pregnancy, the anterior cerebral vesicle is formed, which subsequently divides into 2 lateral ones, which form the basis of the cerebral hemispheres. Next, the cerebral cortex, its convolutions, corpus callosum, basal structures, etc. are formed.
Differentiation of neuroblasts (germinal nerve cells) results in the formation of neurons, which form the gray matter, and glial cells, which make up the white matter. Gray matter is responsible for higher processes of nervous activity. The white matter contains various pathways that connect cerebral structures into a single functioning mechanism. A full-term newborn has the same number of neurons as an adult. But the development of his brain continues, especially intensively in the first 3 months. life. There is an increase in glial cells, branching of neuronal processes and their myelination.
Establishing diagnosis
The following studies are used to diagnose brain malformations:
- computed tomography (determines abnormalities in the gray and white matter of the brain);
- genetic tests;
- magnetic resonance imaging (helps determine the type of disease);
- electroencephalography (helps determine the nature of the electrical activity of the brain).
With polymicrogyria, the cerebral cortex is abnormally thin, and the subarachnoid spaces and ventricles of the brain are enlarged. Defeats
can be located on one or both sides, can be local or extensive. The cortex may appear abnormally thick due to the large number of small convolutions that coalesce to form thick, deep masses.
Pachygyria is determined by MRI results, which show thickening of the cerebral cortex, small gyri, and an underdeveloped Sylvian fissure.
In the prenatal period, it is possible to determine brain malformations using fetal ultrasound. If developmental abnormalities are suspected, they are referred for additional studies (MRI, genetic analysis of candidate genes).
The earliest period at which these pathologies can be determined is the twentieth week of pregnancy.
Epidemiology
Most cases are sporadic, some are X-linked recessive (Xq28). Women experience relatively mild cognitive impairment and subsequently develop epilepsy. In the case of boys, spontaneous termination of pregnancy is observed, usually due to malformations of the cardiovascular system. Those who survive have severe disabilities.
Clinical picture
Most often, subependymal heterotopia is associated with epilepsy and developmental delay.
Pathology
Like other types of heterotopias, this type is the result of impaired neuronal migration. In some cases, the cause of the development of subependymal heterotopia is a violation of cell proliferation.
Gray matter nodules consist of clusters of neurons and glial cells. It is interesting to note that they are most common on the right side, presumably due to the later migration of neuroblasts on the right side.
In X-linked cases, mutations are observed in the gene for filamin-1, a protein that cross-links intracellular actin. In addition, filamin-1 also plays an important role in vascular development.
Diagnostics
MRI is the method of choice, although periventricular heterotopia is visible on CT and ultrasound (if very large).
Ultrasound
Subependymal SV nodules are usually hyperechoic compared with normal white matter, and they may protrude into the ventricular lumen (the wavy edge of the ventricle).
CT
On CT, subependymal heterotopia appears as a non-calcified area of tissue that does not accumulate contrast material, similar in density to normal gray matter, around the lateral ventricles.
MRI
Causes of brain development abnormalities
Failures can occur at various stages of brain formation. If they occur in the first 6 months. pregnancy, they can lead to a decrease in the number of formed neurons, various disorders in differentiation, and hypoplasia of various parts of the brain. At a later date, damage and death of normally formed cerebral substance may occur. The most significant reason for such failures is the influence on the pregnant woman’s body and the fetus of various harmful factors that have a teratogenic effect. The occurrence of an anomaly as a result of monogenic inheritance occurs only in 1% of cases.
The most influential cause of brain defects is considered to be an exogenous factor. Many active chemical compounds, radioactive contamination, and certain biological factors have a teratogenic effect. Of no small importance here is the problem of pollution of the human environment, which causes toxic chemicals to enter the body of a pregnant woman. In addition, various embryotoxic effects may be associated with the lifestyle of the pregnant woman herself: for example, smoking, alcoholism, drug addiction. Dysmetabolic disorders in a pregnant woman, such as diabetes mellitus, hyperthyroidism, etc., can also cause cerebral abnormalities in the fetus. Many medications that a woman can take in the early stages of pregnancy, unaware of the processes occurring in her body, also have a teratogenic effect. Infections suffered by a pregnant woman or intrauterine infections of the fetus have a powerful teratogenic effect. The most dangerous are cytomegaly, listeriosis, rubella, and toxoplasmosis.
genetics
GPR56 is grouped in the B family of GPCRs. This HVHF group has long N ends characterized by an extracellular “cysteine field” and hydrophilic, potentially mucin-rich. The cysteine window contains four conserved cysteines and two tryptophans located in specific modes (C-x2-W-x6-16-W-x4-C-x10-22-CxC) immediately upstream of the first transmembrane domain and serves as a cleavage site in some members of this group of G-protein receptors.
Inheritance mode
Parents of the proband
- The parents of the affected person are obligate heterozygotes and therefore have one mutant allele.
- Heterozygotes (carriers) have no symptoms.
Sibling proband
- In concept, each brother of an affected person has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Once a risk group relative is known to be unaffected, his/her carrier risk is 2/3.
- Heterozygotes (carriers) are asymptomatic.
Proband's offspring
- The proband's offspring are obligate heterozygotes and will therefore carry one mutant allele.
- In highly related populations, the offspring of an individual with a GPR56 linked BFPP and a reproductive partner who is a carrier of the GPR56 linked BFPP has a 50% chance of inheriting two GPR56 disease-causing alleles and has a BFPP and a 50% chance of being carriers.
Other family members of the proband.
- Each brother of the proband's parents is at 50% risk of becoming a carrier
Types of brain development abnormalities
Hemorrhoids kill the patient in 79% of cases
Anencephaly
- absence of the brain and acrania (absence of skull bones). The place of the brain is occupied by connective tissue growths and cystic cavities. May be covered with skin or bare. The pathology is incompatible with life.
Encephalocele
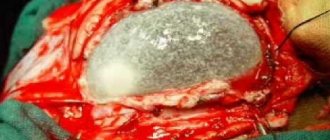
- prolapse of cerebral tissues and membranes through a defect in the bones of the skull, caused by its non-fusion. As a rule, it is formed along the midline, but it can also be asymmetrical. A small encephalocele may mimic a cephalohematoma. In such cases, skull radiography helps determine the diagnosis. The prognosis depends on the size and content of the encephalocele. If the protrusion is small in size and there is ectopic nerve tissue in its cavity, surgical removal of the encephalocele is effective.
Microcephaly
- a decrease in the volume and weight of the brain due to its underdevelopment. Occurs with a frequency of 1 case per 5 thousand newborns. Accompanied by a reduced head circumference and a disproportionate ratio of the facial/cranial skull with a predominance of the former. Microcephaly accounts for about 11% of all cases of mental retardation. With severe microcephaly, idiocy is possible. Often there is not only mental retardation, but also a lag in physical development.
Macrocephaly
- increase in brain volume and mass. Much less common than microcephaly. Macrocephaly is usually combined with disorders of brain architecture and focal heterotopia of the white matter. The main clinical manifestation is mental retardation. Convulsive syndrome may occur. Partial macrocephaly occurs with enlargement of only one of the hemispheres. As a rule, it is accompanied by asymmetry of the cerebral part of the skull.
Cystic cerebral dysplasia
- characterized by multiple cystic cavities of the brain, usually connected to the ventricular system. Cysts can vary in size. Sometimes they are localized only in one hemisphere. Multiple brain cysts manifest as epilepsy that is resistant to anticonvulsant therapy. Single cysts, depending on their size, may have a subclinical course or be accompanied by intracranial hypertension; their gradual resorption is often noted.
Holoprosencephaly
- absence of separation of the hemispheres, as a result of which they are represented by a single hemisphere. The lateral ventricles are formed into a single cavity. Accompanied by severe dysplasia of the facial skull and somatic defects. Stillbirth or death occurs on the first day.
Agiriya
(smooth brain, lissencephaly) - underdevelopment of the gyri and severe violation of the architectonics of the cortex. Clinically manifested by a severe disorder of mental and motor development, paresis and various forms of seizures (including West syndrome and Lennox-Gastaut syndrome). Usually ends in death in the first year of life.
Pachygyria
- enlargement of the main convolutions in the absence of tertiary and secondary ones. Accompanied by shortening and straightening of the grooves, a violation of the architectonics of the cerebral cortex.
Micropolygyria
- The surface of the cerebral cortex is represented by many small convolutions. The bark has up to 4 layers, whereas the normal bark has 6 layers. May be local or diffuse. The latter, polymicrogyria, is characterized by plegia of facial, masticatory and pharyngeal muscles, epilepsy with onset in the 1st year of life, and oligophrenia.
Hypoplasia/aplasia of the corpus callosum
. Often occurs in the form of Aicardi syndrome, described only in girls. Characteristic are myoclonic paroxysms and flexion spasms, congenital ophthalmic defects (colobomas, scleral ectasia, microphthalmos), multiple chorioretinal dystrophic foci detected by ophthalmoscopy.
Focal cortical dysplasia
(FCD) - the presence in the cerebral cortex of pathological areas with giant neurons and abnormal astrocytes. Favorite locations are the temporal and frontal areas of the brain. A distinctive feature of epileptic seizures during FCD is the presence of short-term complex paroxysms with rapid generalization, accompanied in their initial phase by demonstrative motor phenomena in the form of gestures, stomping in one place, etc.
Heterotopias
- accumulations of neurons that, at the stage of neuronal migration, were delayed along their path to the cortex. Heterotopions can be single or multiple, have a nodular or ribbon form. Their main difference from tuberous sclerosis is the lack of the ability to accumulate contrast. These anomalies of brain development are manifested by episyndrome and mental retardation, the severity of which directly correlates with the number and size of heterotopions. With single heterotopia, epileptic seizures, as a rule, debut after 10 years of age.
Characteristic symptoms
The following symptoms are characteristic of polymicrogyria:
- severe developmental delays;
- brain hypoplasia;
- disorders in the facial muscles, as well as the muscles responsible for swallowing, chewing, and tongue movements;
- mental disabilities of varying severity;
- quadriparesis and hemiparesis of the limbs;
- movement disorders;
- breathing complications;
- convulsions;
- epilepsy;
- pseudobulbar syndrome;
- arthrogryposis;
- cerebral paralysis.
Pachygyria manifests itself before the age of 2 years. Its main features are:
- delayed physical and motor development;
- epilepsy;
- hypotension (decreased muscle tone);
- mental retardation;
- infantile spasms;
- microcephaly;
- low muscle control;
- difficulty swallowing, problems eating.
Agyria manifests itself by 3-5 months of a child’s life, less often by 8-9 months; the main symptom of the pathology is expressed in the rapid lag of the child in physical and mental development.
The anomaly may also manifest itself with the following symptoms:
- microcephaly (proportionally small head size);
- growth retardation, speech and motor development;
- general lethargic condition of the child;
- large distance between the eyes, disproportionately small eyes, thin upper lip, high forehead, short nose, low-set ears;
- epilepsy, convulsive syndrome;
- mental retardation;
- increased muscle tone, due to the lack of inhibitory control of the central nervous system over the neurons of the anterior horns of the spinal cord;
- increased distance between the lungs and kidneys;
- rapidly developing dementia;
- lack of control over the pelvic organs (enuresis, fecal incontinence);
- muscular dystrophy.
Cross-sectional photo of the brain and MRI of a child diagnosed with agyria
Diagnosis of brain development abnormalities
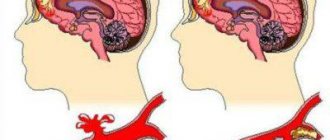
Severe brain abnormalities can often be diagnosed by visual examination. In other cases, cerebral anomaly can be suspected by cerebral retardation, muscle hypotonia in the neonatal period, and the occurrence of convulsive syndrome in children in the first year of life. The traumatic or hypoxic nature of brain damage can be excluded if there is no history of birth trauma of the newborn, fetal hypoxia or asphyxia of the newborn. Prenatal diagnosis of fetal malformations is carried out by screening ultrasound during pregnancy. Ultrasound in the first trimester of pregnancy can prevent the birth of a child with severe cerebral anomaly.
One of the methods for identifying brain defects in infants is neurosonography through the fontanel. Much more accurate data in children of any age and in adults is obtained using MRI of the brain. MRI allows you to determine the nature and localization of the anomaly, the size of cysts, heterotopias and other abnormal areas, and carry out differential diagnosis with hypoxic, traumatic, tumor, and infectious brain lesions. Diagnosis of convulsive syndrome and selection of anticonvulsant therapy is carried out using EEG, as well as prolonged EEG video monitoring. If there are familial cases of cerebral anomalies, consultation with a geneticist with genealogical research and DNA analysis may be useful. In order to identify combined anomalies, examination of somatic organs is carried out: ultrasound of the heart, ultrasound of the abdominal cavity, radiography of the chest organs, ultrasound of the kidneys, etc.
Provoking factors
The main reason for the development of polymicrogyria, pachygyria and agyria is the incorrect process of neuronal migration, which is caused by:
- genetic disorders;
- viral infections;
- insufficient blood supply to the child’s brain during the prenatal period (2nd trimester of pregnancy).
Polymicrogyria can be caused by cytomegalovirus infection, insufficient oxygen saturation of the placenta, and mutations occur in the genes COL18A1 (21q22.3), PAX6 (11p13), K1AA1279 (10q22.1), TUBB2B (6p25), EOMES (3p21.3-p21.2 ), RAB3GAP1 (2q21.3), SRPX2 (Xq21.33-q23).
Agyria can occur due to viral infections that penetrate the uterus or fetus during the first trimester, insufficient blood supply to the fetal brain in the first months of pregnancy. Among the genetic factors are mutations in the RELN gene (7q22), genes of the X chromosome, and 17 chromosomes.
In pachygyria, mutations occur in the LIS1 (17p13.3) and DCX (Xq22.3-q23) genes.
Treatment of brain development abnormalities
Treatment of brain malformations is predominantly symptomatic, carried out by a pediatric neurologist, neonatologist, pediatrician, and epileptologist. In the presence of convulsive syndrome, anticonvulsant therapy is carried out (carbamazepine, levetiracetam, valproate, nitrazepam, lamotrigine, etc.). Since epilepsy in children, which accompanies abnormalities of brain development, is usually resistant to anticonvulsant monotherapy, a combination of 2 drugs is prescribed (for example, levetiracetam with lamotrigine). For hydrocephalus, dehydration therapy is carried out, and shunt operations are used when indicated. In order to improve the metabolism of normally functioning brain tissue, to some extent compensating for the existing congenital defect, it is possible to conduct a course of neurometabolic treatment with the administration of glycine and vitamins. B, etc. Nootropic drugs are used in treatment only in the absence of episyndrome.
For moderate and relatively mild cerebral anomalies, neuropsychological correction, classes for the child with a psychologist, comprehensive psychological support for the child, children's art therapy, and education for older children in specialized schools are recommended. These techniques help to instill self-care skills, reduce the severity of mental retardation and, if possible, socially adapt children with cerebral defects.
The prognosis is largely determined by the severity of the cerebral anomaly. An unfavorable symptom is the earlier onset of epilepsy and its resistance to therapy. The prognosis is complicated by the presence of concomitant congenital somatic pathology.