Аномалии развития головного мозга
— результат происходящих во внутриутробном периоде нарушений формирования отдельных церебральных структур или головного мозга в целом. Зачастую имеют неспецифическую клиническую симптоматику: преимущественно эпилептический синдром, задержку психического и умственного развития. Тяжесть клиники напрямую коррелирует со степенью поражения головного мозга. Диагностируются антенатально при проведении акушерского УЗИ, после рождения — при помощи ЭЭГ, нейросонографии и МРТ головного мозга. Лечение симптоматическое: противоэпилептическое, дегидратационное, метаболическое, психокоррегирующее.
Аномалии развития головного мозга
Аномалии развития головного мозга — пороки, заключающиеся в аномальных изменениях анатомического строения церебральных структур. Выраженность неврологической симптоматики, сопровождающей церебральные аномалии, значительно варьирует. В тяжелых случаях пороки являются причиной антенатальной гибели плода, они составляют до 75% случаев внутриутробной смерти. Кроме того, тяжелые церебральные аномалии обуславливают около 40% случаев гибели новорожденного. Сроки манифестации клинических симптомов могут быть различны. В большинстве случаев церебральные аномалии проявляются в первые месяцы после рождения ребенка. Но, поскольку формирование головного мозга длится до 8-летнего возраста, целый ряд пороков дебютируют клинически после 1-го года жизни. Более чем в половине случаев церебральные пороки сочетаются с пороками соматических органов: врожденными пороками сердца, сращением почек, поликистозом почек, атрезией пищевода и пр. Пренатальное выявление церебральных аномалий является актуальной задачей практической гинекологии и акушерства, а их постнатальная диагностика и лечение — приоритетными вопросами современной неврологии, неонатологии, педиатрии и нейрохирургии.
Формирование головного мозга
Построение нервной системы плода начинается буквально с первой недели беременности. Уже к 23-му дню гестации заканчивается образование нервной трубки, неполное заращение переднего конца которой влечет за собой серьезные церебральные аномалии. Примерно к 28-му дню беременности образуется передний мозговой пузырь, в последующем разделяющийся на 2 боковых, которые ложатся в основу полушарий мозга. Далее образуется кора головного мозга, его извилины, мозолистое тело, базальные структуры и т. д.
Дифференцировка нейробластов (зародышевых нервных клеток) приводит к образованию нейронов, формирующих серое вещество, и глиальных клеток, составляющих белое вещество. Серое вещество отвечает за высшие процессы нервной деятельности. В белом веществе проходят различные проводящие пути, связывающие церебральные структуры в единый функционирующий механизм. Рожденный в срок новорожденный имеет такое же число нейронов, как и взрослый человек. Но развитие его мозга продолжается, особенно интенсивно в первые 3 мес. жизни. Происходит увеличение глиальных клеток, разветвление нейрональных отростков и их миелинизация.
Постановка диагноза
Для диагностики мальформаций головного мозга применяют такие исследования:
- компьютерная томография (определяет нарушения в сером и белом веществе головного мозга);
- генетические анализы;
- магнитно резонансная томография (помогает определить тип заболевания);
- электроэнцефалография (помогает определить характер электрической активности мозга).
При полимикрогирии кора головного мозга аномально тонкая, увеличены субарахноидальные пространства и желудочки мозга. Поражения
могут располагаться с одной или двух сторон, могут быть локальными или обширными. Кора может выглядеть аномально толстой из-за большого количества мелких извилин, которые сливаются и образуют толстые, глубокие массы.
Пахигирию определяют по результатам МРТ, на которых видны утолщения коры головного мозга, небольшие извилины, и недоразвитая сильвиевая борозда.
В пренатальный период определить мальформации мозга возможно при помощи УЗИ плода. При подозрениях на аномалии в развитии направляют на дополнительные исследования (МРТ, генетический анализ генов-кандидатов).
Самым ранним сроком, на котором можно определить данные патологии, является двадцатая неделя беременности.
Эпидемиология
Большинство случаев являются спорадическими, некоторые являются рецессивными, сцепленными с Х-хромосомой (Хq28). У женщин наблюдается относительно мягкие когнитивные нарушения, впоследствии развивается эпилепсия. В случае мальчиков наблюдается спонтанное прерывание беременности, обычно из-за пороков развития сердечно-сосудистой системы. У выживших – тяжелая инвалидность.
Клиническая картина
Чаще всего субэпендимальная гетеротопия ассоциирована с эпилепсией и задержкой развития.
Патология
Как и другие типы гетеротопий данный вид является результатом нарушения нейрональной миграции. В некоторых случаях причиной развития субэпендимальной гетеротопии является нарушение клеточной пролиферации.
Узелки серого вещества состоят из кластеров нейронов и глиальных клеток. Интересно заметить, что они чаще всего встречаются справа, предположительно, из-за более поздней миграции нейробластов с правой стороны.
В случаях, сцепленных с Х-хромосомой, наблюдаются мутации в гене филамина-1, белка, который перекрестно связывает внутриклеточный актин. Кроме того, филамин-1 также играет важную роль в развитии сосудов.
Диагностика
Методом выбора является МРТ, хотя перивентрикулярная гетеротопия видна на КТ и УЗИ (при очень большом размере).
УЗИ
Субэпендимальные узелки СВ обычно гиперэхогенны по сравнению с нормальным белым веществом, а также они могут выступать в просвет желудочков (волнообразный край желудочка).
КТ
На КТ субэпендимальная гетеротопия выглядит как некальцифицированный участок ткани, не накапливающий контрастное вещество, по плотности, схожей с нормальным серым веществом, вокруг боковых желудочков.
МРТ
Причины аномалий развития головного мозга
Сбои могут произойти на различных этапах формирования головного мозга. Если они возникают в первые 6 мес. беременности, то способны приводить к снижению числа сформированных нейронов, различным нарушениям в дифференцировке, гипоплазии различных отделов мозга. В более поздние сроки может возникать поражение и гибель нормально сформировавшегося церебрального вещества. Наиболее весомой причиной подобных сбоев является влияние на организм беременной и на плод, различных вредоносных факторов, обладающих тератогенным действием. Возникновение аномалии в результате моногенного наследования встречается лишь в 1% случаев.
Наиболее влиятельной причиной пороков головного мозга считается экзогенный фактор. Тератогенным эффектом обладают многие активные химические соединения, радиоактивное загрязнение, отдельные биологические факторы. Немаловажное значение здесь имеет проблема загрязнения среды обитания людей, обуславливающая поступление в организм беременной токсических химических веществ. Кроме того, различные эмбриотоксические воздействия могут быть связаны с образом жизни самой беременной: например, с курением, алкоголизмом, наркоманией. Дисметаболические нарушения у беременной, такие как сахарный диабет, гипертиреоз и пр., могут также стать причиной церебральных аномалий плода. Тератогенным действием обладают и многие медикаменты, которые может принимать женщина в ранние сроки беременность, не подозревая о происходящих в ее организме процессах. Мощный тератогенный эффект оказывают инфекции, перенесенные беременной, или внутриутробные инфекции плода. Наиболее опасны цитомегалия, листериоз, краснуха, токсоплазмоз.
генетика
GPR56 группируется в B семейства GPCRs. Эта ХВГФ группа имеет длинные N — концы характеризующихся внеклеточное «цистеин поле» и гидрофильным, потенциально муцин -Rich. Окно цистеина содержит четыре консервативных цистеина и два триптофаны , расположенных в определенных модах (С-x2-W-x6-16-W-x4-С-x10-22-СхС) непосредственно перед первым трансмембранным доменом и служит в качестве сайта расщепления в некоторых членов этой группы G-белком рецепторов.
Режим наследования
Родители пробанда
- Родители больного лица облигатные гетерозиготы и , следовательно , имеют один мутантный аллель.
- Гетерозигот (носители) не имеют симптомов.
Сибсов пробанда
- В концепции, каждый брат пораженного человека имеет 25% шансов быть затронуты, 50% шансов быть бессимптомным носителем, и 25% шанс быть не влияет на и не носитель.
- После того, как группы риска родственный, как известно, не влияет, риск его / ее носительства составляет 2/3.
- Гетерозигот (носители) являются бессимптомными .
Потомство пробанда
- Потомство пробанда облигатные гетерозиготы и, следовательно, будет нести один мутантный аллель.
- В популяциях с высокой степенью родства, потомок человека с GPR56 связанным BFPP и репродуктивным партнером, который является носителем GPR56 связанных BFPP имеет 50% шанс унаследовать две GPR56 болезнетворных аллелей и имеющий BFPP и 50% шансов быть носителями.
Другие члены семьи пробанда .
- Каждый брат родителей пробанда находится на 50% риск стать носителем
Виды аномалий развития головного мозга
Геморрой в 79% случаев убивает пациента
Анэнцефалия
— отсутствие головного мозга и акрания (отсутствие костей черепа). Место головного мозга занято соединительнотканными разрастаниями и кистозными полостями. Может быть покрыто кожей или обнажено. Патология несовместима с жизнью.
Энцефалоцеле
— пролабирование церебральных тканей и оболочек через дефект костей черепа, обусловленный его незаращением. Как правило, формируется по средней линии, но бывает и асимметричным. Небольшое энцефалоцеле может имитировать кефалогематому. В таких случаях определить диагноз помогает рентгенография черепа. Прогноз зависит от размеров и содержимого энцефалоцеле. При небольших размерах выпячивания и наличии в его полости эктопированной нервной ткани эффективно хирургическое удаление энцефалоцеле.
Микроцефалия
— уменьшение объема и массы головного мозга, обусловленное его недоразвитием. Встречается с частотой 1 случай на 5 тыс. новорожденных. Сопровождается уменьшенной окружностью головы и диспропорциональным соотношением лицевого/мозгового черепа с преобладанием первого. На долю микроцефалии приходится около 11% всех случаев олигофрении. При выраженной микроцефалии возможна идиотия. Зачастую наблюдается не только ЗПР, но и отставание в физическом развитии.
Макроцефалия
— увеличение объема головного мозга и его массы. Гораздо менее распространена, чем микроцефалия. Макроцефалия обычно сочетается с нарушениями архитектоники мозга, очаговой гетеротопией белого вещества. Основное клиническое проявление — умственная отсталость. Может наблюдаться судорожный синдром. Встречается частичная макроцефалия с увеличением лишь одного из полушарий. Как правило, она сопровождается асимметрией мозгового отдела черепа.
Кистозная церебральная дисплазия
— характеризуется множественными кистозными полостями головного мозга, обычно соединенными с желудочковой системой. Кисты могут иметь различный размер. Иногда локализуются только в одном полушарии. Множественные кисты головного мозга проявляются эпилепсией, устойчивой к антиконвульсантной терапии. Единичные кисты в зависимости от размера могут иметь субклиническое течение или сопровождаться внутричерепной гипертензией; зачастую отмечается их постепенное рассасывание.
Голопрозэнцефалия
— отсутствие разделения полушарий, в результате чего они представлены единой полусферой. Боковые желудочки сформированы в единую полость. Сопровождается грубыми дисплазиями лицевого черепа и соматическими пороками. Отмечается мертворождение или гибель в первые сутки.
Агирия
(гладкий мозг, лиссэнцефалия) — недоразвитие извилин и тяжелое нарушение архитектоники коры. Клинически проявляется выраженным расстройством психического и моторного развития, парезами и различными формами судорог (в т. ч. синдромом Веста и синдромом Леннокса-Гасто). Обычно заканчивается летальным исходом на первом году жизни.
Пахигирия
— укрупнение основных извилин при отсутствии третичных и вторичных. Сопровождается укорочением и выпрямлением борозд, нарушением архитектоники церебральной коры.
Микрополигирия
— поверхность коры мозга представлена множеством мелких извилин. Кора имеет до 4-х слоев, тогда как в норме кора насчитывает 6 слоев. Может быть локальной или диффузной. Последняя, полимикрогирия, характеризуется плегией мимических, жевательных и глоточных мышц, эпилепсией с дебютом на 1-ом году жизни, олигофренией.
Гипоплазия/аплазия мозолистого тела
. Часто встречается в виде синдрома Айкарди, описанного только у девочек. Характерны миоклонические пароксизмы и сгибательные спазмы, врожденные офтальмические пороки (колобомы, эктазия склеры, микрофтальм), множественные хориоретинальные дистрофические очаги, обнаруживаемые при офтальмоскопии.
Фокальная корковая дисплазия
(ФКД) — наличие в коре головного мозга патологических участков с гигантскими нейронами и аномальными астроцитами. Излюбленное расположение — височные и лобные зоны мозга. Отличительной особенностью эпиприступов при ФКД является наличие кратковременных сложных пароксизмов с быстрой генерализацией, сопровождающихся в своей начальной фазе демонстративными двигательными феноменами в виде жестов, топтания на одном месте и т. п.
Гетеротопии
— скопления нейронов, на этапе нейронной миграции задержавшихся на пути своего следования к коре. Гетеротопионы могут быть единичными и множественными, иметь узловую и ленточную форму. Их главное отличие от туберозного склероза — отсутствие способности накапливать контраст. Эти аномалии развития головного мозга проявляются эписиндромом и олигофренией, выраженность которых прямо коррелирует с числом и размером гетеротопионов. При одиночной гетеротопии эпиприступы, как правило, дебютируют после 10-летнего возраста.
Характерная симптоматика
Для полимикрогирии характерны такие симптомы:
- тяжелые задержки в развитии;
- гипоплазия головного мозга;
- нарушения в лицевых мышцах, а также мышцах отвечающих за глотание, жевания, движения языком;
- умственные отклонения разной степени тяжести;
- квадрипарез и гемипарез конечностей;
- двигательные нарушения;
- осложнения дыхания;
- судороги;
- эпилепсия;
- псевдобульбарный синдром;
- артрогрипоз;
- церебральный паралич.
Пахигирия проявляет себя в возрасте до 2 лет. Основными ее признаками являются:
- задержка физического, двигательного развития;
- эпилепсия;
- гипотония (пониженный мышечный тонус);
- умственная отсталость;
- инфантильные спазмы;
- микроцефалия;
- низкий контроль мышц;
- трудности при глотании, проблемы с приемом пищи.
Агирия проявляется к 3-5 месяцу жизни ребенка, реже к 8-9 месяцу, основной симптом патологии выражается в стремительном отставании ребенка в физическом и умственном развитии.
Также аномалия может проявлять себя следующими признаками:
- микроцефалия (пропорционально небольшой размер головы);
- отставание в росте, в речевом и двигательном развитии;
- общее вялое состояние ребенка;
- большое расстояние между глазами, диспропорционально маленькие глаза, тонкая верхняя губа, высокий лоб, короткий нос, низко расположенные уши;
- эпилепсия, судорожный синдром;
- отставание в умственном развитии;
- повышенный мышечный тонус, из-за отсутствия тормозящего контроля центральной нервной системы над нейронами передних рогов спинного мозга;
- увеличенное расстояние между легкими, почками;
- стремительно развивающееся слабоумие;
- отсутствие контроля над органами малого таза (энурез, недержание кала);
- мышечная дистрофия.
Фото мозга в разрезе и на МРТ ребенка с диагнозом агирия
Диагностика аномалий развития головного мозга
Тяжелые аномалии развития головного мозга зачастую могут быть диагностированы при визуальном осмотре. В остальных случаях заподозрить церебральную аномалию позволяет ЗПР, гипотония мышц в неонатальном периоде, возникновение судорожного синдрома у детей первого года жизни. Исключить травматический или гипоксический характер поражения головного мозга можно при отсутствии в анамнезе данных о родовой травме новорожденного, гипоксии плода или асфиксии новорожденного. Пренатальная диагностика пороков развития плода осуществляется путем скринингового УЗИ при беременности. УЗИ в I триместре беременности позволяет предупредить рождение ребенка с тяжелой церебральной аномалией.
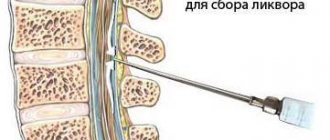
Одним из методов выявления пороков головного мозга у грудничков является нейросонография через родничок. Намного более точные данные у детей любого возраста и у взрослых получают при помощи МРТ головного мозга. МРТ позволяет определить характер и локализацию аномалии, размеры кист, гетеротопий и других аномальных участков, провести дифференциальную диагностику с гипоксическими, травматическими, опухолевыми, инфекционными поражениями мозга. Диагностика судорожного синдрома и подбор антиконвульсантной терапии осуществляется при помощи ЭЭГ, а также пролонгированного ЭЭГ-видеомониторинга. При наличии семейных случаев церебральных аномалий может быть полезна консультация генетика с проведением генеалогического исследования и ДНК-анализа. С целью выявления сочетанных аномалий проводится обследование соматических органов: УЗИ сердца, УЗИ брюшной полости, рентгенография органов грудной полости, УЗИ почек и пр.
Провоцирующие факторы
Основная причина развития полимикрогирии, пахигирии и агирии в неправильном протекании процесса нейрональной миграции, причиной чего являются:
- генетические нарушения;
- вирусные инфекции;
- недостаточное кровоснабжение головного мозга ребенка в пренатальный период (2 триместр беременности).
Полимикрогирия может быть вызвана цитомегаловирусной инфекцией, недостаточным насыщением плаценты кислородом, а мутации происходят в генах COL18A1 (21q22.3), PAX6 (11p13), K1AA1279 (10q22.1), TUBB2B (6p25), EOMES (3p21.3-p21.2), RAB3GAP1 (2q21.3), SRPX2 (Xq21.33-q23).
Агирия может возникнуть из-за вирусных инфекций, которые проникают в матку или в плод в период первого триместра, недостаточного кровоснабжения мозга плода в первые месяцы беременности. Среди генетических факторов можно назвать мутации в гене RELN (7q22), генах X хромосомы, 17 хромосомы.
При пахигирии возникают мутации в генах LIS1 (17p13.3) и DCX (Xq22.3-q23).
Лечение аномалий развития головного мозга
Терапия пороков развития головного мозга преимущественно симптоматическая, осуществляется детским неврологом, неонатологом, педиатром, эпилептологом. При наличии судорожного синдрома проводится антиконвульсантная терапия (карбамазепин, леветирацетам, вальпроаты, нитразепам, ламотриджин и др.). Поскольку эпилепсия у детей, сопровождающая аномалии развития головного мозга, обычно резистентна к противосудорожной монотерапии, назначают комбинацию из 2 препаратов (например, леветирацетам с ламотриджином). При гидроцефалии осуществляют дегидратационную терапию, по показаниям прибегают к шунтирующим операциям. С целью улучшения метаболизма нормально функционирующих мозговых тканей, в какой-то степени компенсирующих имеющийся врожденный дефект, возможно проведение курсового нейрометаболического лечения с назначением глицина, витаминов гр. В и пр. Ноотропные препараты используются в лечении только при отсутствии эписиндрома.
При умеренных и относительно легких церебральных аномалиях рекомендована нейропсихологическая коррекция, занятия ребенка с психологом, комплексное психологическое сопровождение ребенка, детская арт-терапия, обучение детей старшего возраста в специализированных школах. Указанные методики помогают привить навыки самообслуживания, уменьшить степень выраженности олигофрении и по возможности социально адаптировать детей с церебральными пороками.
Прогноз во многом определяется тяжестью церебральной аномалии. Неблагоприятным симптомом выступает ранее начало эпилепсии и ее резистентность к осуществляемой терапии. Осложняет прогноз наличие сочетанной врожденной соматической патологии.