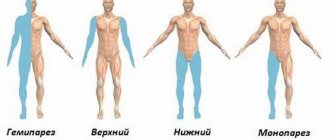
PROGRESSIVE PARALYSIS
(
paralysis progressiva
, synonym:
progressive paralysis of the insane, paralytic dementia, Bayle's disease
) is a late form of neurosyphilis, characterized by impaired mental activity - progressive total dementia.
One of the first cases of undoubted P. p. was described by J. Esquirol. In 1822, A. Bayle identified P. p. as an independent disease, emphasizing that it causes both mental disorders and neurol, disorders associated with hron. inflammation of the arachnoid membrane of the brain. Bayle's statement has met with objections from some researchers. And only in the 50s. 19th century progressive paralysis was recognized as an independent disease.
P. p. suffers approx. 5% of all patients with syphilis (see), and women get sick much less often than men. The disease usually occurs 10-15 years or more after infection. Thus, if we take into account that the period of greatest sexual activity falls by 20-35 years, the age of those affected is most often 35-50 years. When infected with syphilis at a late age (45-60 years), this period is usually shortened.
History of the study
Until the middle of the 20th century, this disease was not considered as an independent nosology; it was mistakenly interpreted as the consequences of epidemic encephalitis, which was widespread at that time.
The fact is that a large number of cases of polymorphic postencephalitic parkinsonism masked rarer pathologies that were considered atypical forms. Progressive supranuclear palsy as an independent neuropathology was identified in 1963-1964. a group of Canadian doctors: neurologists J. Steel and J. Richardson and pathologist J. Olszewski. They described and analyzed 7 cases of neurodegeneration with a characteristic clinical picture. In the USSR, progressive supranuclear palsy was first mentioned in 1980 by doctors at the Clinic of Nervous Diseases of the Moscow Medical Academy. THEM. Sechenov, who observed two patients.
Subsequently, the disease continued to be studied and was identified in domestic and world classifications as a separate nosological unit. In ICD-10, progressive supranuclear palsy is classified as a disease of the nervous system (section of extrapyramidal and other motor disorders, subsection of other degenerative diseases of the basal ganglia), coded G23.1.
Symptoms of PSP
Progressive supranuclear palsy is characterized by a nonspecific clinical onset. Symptoms of this period include unusual fatigue, decreased performance, cephalgia, dizziness, low mood, narrowing of interests, sleep disorders, including insomnia at night and hypersomnia during the day. Subsequently, symptoms of akinetic-rigid parkinsonism appear. Postural tremor is absent in most patients. Muscle rigidity is expressed primarily in the axial muscles—the muscles that run along the cervical spine, connecting it to the skull. Patients complain of stiffness in the neck and back. Increased tone in the posterior muscles of the neck leads to a typical “proud” position of the patient’s head. Parkinsonian ataxia is characteristic, caused by a disorder of coordination of the position of the trunk and lower extremities relative to the center of gravity. Difficulty maintaining balance while walking leads to frequent falls backwards.
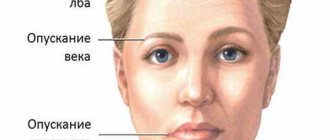
A distinctive feature of PSP is ophthalmoplegia, which occurs on average 2-3 years after the onset of the disease. Against the background of slow movement of the eyeballs, gaze paralysis occurs in the vertical plane, the patient cannot lower his eyes down. Due to infrequent blinking, the patient feels discomfort and burning in the eyes. Possible blurred vision, convergence disorder, blepharospasm. Progressive supranuclear ophthalmoparesis is accompanied by limitation of gaze down and up, and over time can lead to oculomotor disorders in the horizontal plane. With the development of complete ophthalmoplegia, retraction of the upper eyelids is formed, which gives the face a surprised expression.
In the clinical picture of PSP, pseudobulbar manifestations appear relatively early: dysarthria, dysphagia, forced crying or laughter. Changes occur in the personal-emotional sphere, patients become withdrawn, apathetic, demotivated, and indifferent. Cognitive impairment in most cases occurs at the height of the disease, in 10-30% of cases - at the onset stage. Characterized by intellectual decline, disorders of abstract thinking and memory, visuospatial apraxia, elements of agnosia. Dementia is observed in 60% of patients with a 3-year history of the disease.
The disease begins with nonspecific, slowly increasing symptoms, similar to neurasthenic manifestations. Patients complain of decreased performance, unusual fatigue, irritability, headaches, memory impairment, and sleep disturbances. Over time, the patient’s relatives notice changes in his personality: indifference to the problems of others, loss of a sense of tact, behavior beyond ethical standards. Patients begin to make gross mistakes in their usual professional activities, and as symptoms progress, they lose the ability to recognize them. Sleep disorders worsen: hypersomnia occurs during the day and insomnia at night. Eating disorders occur (anorexia, bulimia). Speech speeds up or slows down excessively. Criticism of one's behavior decreases.
At the stage of development of the disease, progressive paralysis is accompanied by worsening personality and behavioral disorders. Inappropriate behavior, ridiculous actions, and flat jokes are revealed. Mental disorders are characterized by polymorphism, euphoria, depression, delusions, hallucinations, and depersonalization are found. In speech, difficulties arise in pronouncing complex words, then dysarthria develops - speech becomes slurred, blurred, with the omission of individual sounds.
The gait is unsteady, loose, the handwriting is uneven, dysgraphia with missing letters is noted. Fainting and epileptic seizures are observed (usually of the Jacksonian type). Appetite disturbances cause weight loss or weight gain. Trophic disorders appear: decreased skin turgor, brittle nails, mild infection with the development of boils and abscesses. Characterized by increased fragility of bones, degenerative changes in the heart (cardiomyopathy) and liver (hepatosis).
During the period of dementia, there is an increase in dementia up to the point of insanity. The patient stops serving himself, interacting with others, and voluntarily controlling the function of the pelvic organs. Refusal to eat leads to significant weight loss. Dysphagia, a swallowing disorder, is sometimes noted. Numerous trophic ulcers form, and increasing bone fragility causes fractures.
Patients need etiotropic and psychocorrective therapy. The therapeutic effect of high body temperature was experimentally discovered. Modern treatment includes four main components:
- Pyrotherapy. Previously, it was carried out by introducing malaria pathogens followed by antimalarial therapy. Currently, pyrogenic drugs are used.
- Antibacterial therapy. Penicillin antibiotics, ceftriaxone, quinine iodobismuthate, and bismuth nitrate have an antitreponemal effect. Etiotropic therapy begins with large doses and continues for 2-3 weeks. At the same time, probiotics, multivitamin preparations, and, if necessary, hepatoprotectors are prescribed.
- Psychotropic treatment. Indicated in the second phase of Bayle's disease. Individual selection of pharmaceuticals (sedatives, antipsychotics, antipsychotics, antidepressants) is carried out in accordance with the symptoms.
- Neurotropic therapy. Aimed at improving metabolic processes in the brain and restoring its functions. Includes pharmaceuticals with vascular, nootropic, and neurometabolic effects. At the stage of dementia, neurotropic treatment does not have a significant effect.
Prevalence
The disease is based on the progressive degeneration of neurons, resulting from the accumulation of pathological protein in them.
According to modern medical statistics, progressive supranuclear palsy is the cause of 4-7% of cases of diagnosed parkinsonism. But even now, some patients with this disease are misdiagnosed, especially in the early stages. The overall prevalence of progressive supranuclear palsy in the population averages 5 cases per 100 thousand population, from 1.4 to 6.4 cases in different countries.
The disease is detected mainly in people of the older age group; even hereditary forms usually appear at the age of 50 years.
Clinical picture
Rest tremor is not typical for progressive supranuclear palsy.
All symptoms of progressive supranuclear palsy are combined into several groups:
- Oculomotor disorders in the form of gaze paralysis and a number of other symptoms, accompanied by retraction (lifting) of the upper eyelids with the formation of a characteristic “surprised” facial expression.
- Parkinsonism (akinetic-rigid form). Moreover, extrapyramidal disorders in the classic course of progressive supranuclear palsy have a number of features that allow for correct differential diagnosis. Characterized by the predominance of rigidity of the muscles of the neck and shoulder girdle with the formation of a characteristic “proud” posture, bradykinesia (slowness of movements), symmetry of disorders even in the initial stages, and the early appearance of postural instability. These extrapyramidal symptoms are not corrected by antiparsinsonian drugs. Rest tremor, falls, and obvious autonomic and pelvic disorders are not typical.
- Walking disorders, usually of the subcortical astasia type with a pronounced influence of postural instability. In this case, the length of the step, the area of support and the initiation of movements initially do not change, and the friendly movements of the arms and legs are preserved. People with progressive supranuclear palsy, already in the early stages of the disease, easily lose stability when turning, changing speed, pushing, or walking on an inclined surface. During the first year of the disease, falls backward occur, without attempts to maintain balance.
- Cognitive impairment, with fairly rapid development of dementia of the frontal-subcortical type. Speech becomes impoverished, the ability to abstract and generalize is lost, apathy, field behavior, low speech activity, and echopraxia are characteristic.
- Pseudobulbar syndrome caused by damage to the frontal cortex and the regulatory pathways coming from it. Dysarthria (unclear sound pronunciation), dysphagia (swallowing disorders, with a preserved or even increased pharyngeal reflex), symptoms of oral automatism, forced laughter and crying develop early.
Progressive supranuclear palsy is not characterized by illusions, hallucinatory-delusional syndrome, qualitative and quantitative disturbances of consciousness, and severe affective disorders.
Atypical clinical forms of progressive supranuclear palsy are also possible: with a predominance of parkinsonism and the appearance of asymmetrical dystonia of the limbs, with a debut in the form of rapidly increasing cognitive impairment, with a predominance of primary progressive aphasia.
P. p. is a serious disease of the whole organism, and its most striking manifestations are disturbances in mental activity. The main syndrome is progressive total dementia (see): the intellect suffers severely, disorders of judgment appear early, criticism and especially self-criticism disappear. There is no awareness of the disease, memory sharply decreases, and confabulations occur (see Confabulosis).
Manifestations of dementia intensify due to the often observed euphoria (see Psychoorganic syndrome). Neurol. the symptoms consist of speech disorders, primarily in violation of articulation - dysarthria (see). Speech becomes unclear, slurred, especially when pronouncing long words, the patient skips or rearranges syllables, and does not pronounce the endings of words.
Handwriting becomes uneven, individual letters and syllables drop out of words. The timbre of the voice changes, it becomes dull. The patient's face is expressionless and mask-like, because the innervation of the facial muscles is disrupted, and blepharoptosis occurs (see Ptosis). Tendon reflexes are often increased and uneven, except in cases of taboparalysis (see).
Based on psychopathol. manifestations, four stages of the disease are distinguished: latent (from infection with syphilis to manifestations of P. p.), the stage of initial manifestations, the stage of full development of the disease and the stage of marasmus (see). In the latent stage, headaches, dizziness, fainting may occur, and in some cases characteristic changes in the cerebrospinal fluid are observed (see).
The stage of initial manifestations is characterized by increased fatigue, irritability, and weakness. Patients complain of loss of strength and decreased performance, although they can still perform their usual work to some extent. Previously, such conditions, due to their external resemblance to neurotic symptoms, were incorrectly called preparalytic neurasthenia.
In some cases, at the stage of initial manifestations, depressive and delusional disorders are observed - anxious depression with hypochondriacal statements, anxious-agitated depression, delusional ideas of jealousy, persecution, poisoning; as the symptoms of dementia increase, these endoform disorders disappear.
Memory loss is detected very early. Certain actions indicate a violation of criticism. The sphere of desires is upset, patients become gluttonous and erotic. The increase in these disorders indicates the transition of the disease to the stage of full development, the edges manifest themselves in different wedges. forms.
The expansive, or classic, form (previously widespread) is more common in men. It is characterized by the presence of manic excitement with the manifestation of anger, grandiose delusions of grandeur (see Delirium). The dementia form is characterized by increasing dementia against the background of inactive euphoria. In the depressive form, a depressed mood develops, often with anxiety and a desire for suicide (see.
Depressive syndromes), often there is an absurd hypochondriacal delusion of nihilistic content. The circular form, first described by S.S. Korsakov, occurs with alternating states of excitement and depression. The hallucinatory-paranoid form is characterized by the development of paranoid syndrome (see) with predominantly auditory hallucinations and delusions of persecution.
In the catatonic form, a stuporous state occurs (see) with phenomena of mutism and negativism (see Catatonic syndrome). In the stage of insanity, conscious activity ceases, speech disappears, patients make inarticulate sounds, and cannot stand or move. At this stage they die from an intercurrent disease.
Along the way, a particularly malignant agitated form is distinguished (galloping paralysis) with sharp motor excitation and disturbance of consciousness of the amentive type and the so-called. stationary paralysis, in which there is a slow course with a gradual decrease in intelligence and lethargy.
Atypical forms of P. p. are youthful and senile P. p., as well as Lissauer’s palsy and taboparalysis (see). Juvenile P. p. develops on the basis of congenital syphilis; usually begins at the age of 10-15 years. Sometimes it is preceded by signs of congenital syphilis, in other cases it occurs in children who were previously considered healthy.
Most often occurs in the form of dementia; Local symptoms are often observed, for example, optic nerve atrophy. Senile P. p. occurs over the age of 60 years and is characterized, first of all, by a long latent stage (up to 40 years). Wedge, the picture resembles senile dementia (see) with severe memory disorders; sometimes the disease occurs like Korsakov's syndrome (see).
Lissauer's palsy and taboparalysis are characterized by a relatively slow progression of dementia. With Lissauer's palsy, there is a tendency to local damage to the brain, mainly the parietal lobes, with the development of aphasia (see), agnosia (see), apraxia (see), apoplectiform and epileptiform seizures.
Differential diagnosis
Differential diagnosis of manic progressive paralysis and a manic attack of manic-depressive psychosis is based on the presence of somatic symptoms, on the discrepancy between the degree of arousal and colossal nonsense, and most importantly on the absurdity of delusional ideas. A manic patient rather seems to be playing around, calling people by names other than theirs; the paralytic believes in his own fictions.
The first attack of the disease in adulthood requires great care in evaluation, even when there are no paralytic symptoms; Today they are not there, tomorrow they will be. In any case, delusional ideas are almost always clearly expressed in the manic form of progressive paralysis.
The same principles should be followed in the differential diagnosis of melancholic attacks.
It is not always easy to distinguish progressive paralysis from schizophrenia, especially if there is only a vague state of excitement with a meaningless change of ideas, a state resembling confusion, but without any specific coloring. The catatonic signs observed in this case (they can be difficult to accurately analyze in this state of the patient) do not in themselves prove the presence of catatonia; inhibition of affect and the absence of mental and affective connection with the outside world are more evident. History can often confuse matters further, since many paralytics are psychopaths even in a healthy state. In these cases, the decisive role will be played by whether it will be possible to prove (or categorically refute) that the patient has noticed a paralytic change in character from a certain moment.
To distinguish it from epilepsy, you only need to keep in mind the characteristic features of both diseases; however, when epileptic seizures first appear in middle age, they do not have a definite character in either direction; in such cases, the diagnosis remains unclear for some time.
Due to the tendency of paralytics to excesses, it is sometimes very difficult to distinguish progressive paralysis from alcoholic forms. Korsakoff's disease, being an organic psychosis, has a number of mental symptoms in common with progressive paralysis, but the difficulty is eliminated by neuritis, the absence of specific paralytic symptoms (sometimes pupillary symptoms are observed), and at times the onset of the disease. It should also be noted that neuritis is not always clearly expressed.
The distinction from brain tumors is easy for obvious reasons; however, it should be noted that slow-growing, infiltrating gliomas are encountered, which clinically present a picture of a simple form of progressive paralysis with barely noticeable local symptoms.
Progressive paralysis differs from other organic psychoses in the presence of somatic symptoms (including the composition of the cerebrospinal fluid); however, even when they are not present, a diagnosis of progressive paralysis can be made without great risk on the basis of paralytic delusions of grandeur alone.
An important task is to establish the exact boundary between progressive paralysis and neurasthenia.
All doubts about the diagnosis are dispelled by special reactions of the blood and cerebrospinal fluid to syphilis. The most famous of these is the Wassermann reaction (RW).
Pathogenesis: what happens in the brain?
Progressive paralysis without appropriate treatment usually leads to death within several years. The disease almost always creeps up gradually, this also applies to those cases when an acute attack suddenly makes the disease obvious to others.
Already 10 years before the outbreak of the disease, one can sometimes detect both pupillary disorders and a change in character; Often, for years, only neurasthenic symptoms are observed, behind which lies a formidable disease.
Progressive paralysis, once manifested, can give remissions, and so deep that they simulate recovery (with simple forms of dementia this almost never happens).
The alternation of individual symptoms does not obey any law. In the period of precursors, that is, when the disease has not yet been recognized, the most common are pupillary symptoms, handwriting disorder and character changes. The disease can debut with a neurasthenic symptom, paralytic attack or other somatic symptom just as well as with a sharp mental syndrome (criminal or absurd act).
The most common early symptoms include pupillary disorders, loss of knee reflexes (when complicated by tabes dorsalis), irritability, fears, exaggerated entrepreneurship, or, conversely, unusual weakness of will, twilight states for several minutes and even hours, attacks of dizziness, fainting, transient double vision, periodic difficulty speaking, sleep disturbance and especially neurasthenic syndrome.
The initial stage in different forms is more or less the same; the difference is only in the state of the affective sphere (most are euphoric) and in the presence or absence of eretic states.
Death in uncomplicated cases occurs from exhaustion. Paralysis of the bladder and bedsores give rise to all kinds of infections; paralysis of the swallowing and respiratory muscles leads to pneumonia or asphyxia. A paralytic seizure itself can also result in death. Some patients become victims of accidents due to their helplessness, both motor and mental; in depressive cases, rarely in simple ones, the disease sometimes ends in the patient’s suicide.
The causes of progressive supranuclear palsy are not reliably known. This disease is not associated with any infections, previous injuries or exposure to external adverse factors.
Progressive supranuclear palsy is considered a sporadic pathology (appearing randomly in the population). At the same time, since 1995, information has been published on the presence of rare familial cases of the disease with an autosomal dominant type of inheritance. This variant of the disease is associated with a heterozygous mutation of the gene encoding tau protein and located on 17q21.31.
The development of symptoms of progressive supranuclear palsy is associated with irreversible and steadily increasing degeneration of neurons in certain areas of the brain. The basis of this destructive process is the excessive intracellular accumulation of neurofibrillary tangles and neuropil threads that have lost their structure. They disrupt the functioning of neurons and promote their premature apoptosis (programmed self-destruction).
Neurofibrillary tangles in the cytoplasm of brain neurons are formed by a special τ protein (tau protein), which is in a pathological hyperphosphorylated state. Normally, it is attached to tubulin microtubules and is responsible for their polymerization and stabilization of microtubules and fixation of some intracellular enzymes.
The main functions of normal tau protein include:
- participation in the processes of maintaining the neuronal cytoskeleton (nerve cell framework);
- formation and elongation of axonal processes;
- restoration of neurons after damage;
- regulation of intracellular transport of vesicles (cytoplasmic vesicles) with synthesized neuropeptides.
Hyperphosphorylated tau protein is no longer able to maintain the microtubule structure. They disintegrate, and the protein that has become abnormal forms filaments (tubules) of irregular shape, which gather in the cytoplasm into neurofibrillary tangles. In the affected cell, biochemical contact with other neurons is disrupted, the ability to form and maintain axonal connections is lost, the cytoskeleton becomes unstable, and the lifespan is significantly reduced. Such neurodegeneration is irreversible and progressive, gradually spreading from the characteristic primary zones to the entire brain.
Neurofibrillary tangles in neurons are formed not only during progressive supranuclear palsy. Similar degenerative changes in the brain are also found in Alzheimer's disease, corticobasal degeneration, frontotemporal dementia and some other, more rare diseases.
The specially created Reisensburg Working Group for Tauopathies With Parkinsonism is studying various tauopathies, developing issues of their differential diagnosis and clarifying nosological criteria.
In 1913, H. Noguchi proved the syphilitic etiology of P. p. by discovering treponema pallidum in brain tissue.
Pathogenesis has not been studied enough. K. Levaditi, based on observations when both spouses or several persons infected with syphilis from the same source fell ill with syphilis, suggested the existence of a special neurotropic treponema. which, however, has not been proven. Perhaps disturbances in the reactivity of the body (see) with sensitization of brain tissue play a role in pathogenesis, as a result of which in some cases treponemes penetrate into the brain tissue.
Treatment
Pyrotherapy is indicated (see) - inf. therapy and pyrogenic substances (see) in combination, as a rule, with antibiotic therapy. Back in 1845, V.F. Sabler noted the beneficial effect of a number of febrile diseases on the course of psychoses. Priority in inf. therapy of psychosis belongs to A. S. Rosenblum, who treated mentally ill patients with relapsing fever vaccination. In 1917, the Viennese psychiatrist J. Wagner-Jauregg proposed treating patients with P. p. with malaria vaccinations. The method has become widespread; it consists of injecting subcutaneously into a patient P. a p. of blood taken from a patient with three-day malaria. The incubation period lasts 4-20 days, the first attacks are similar to three-day malaria, and then, as a rule, are observed daily. After 10-12 attacks, they are stopped by taking quinine hydrochloride. Inf. therapy is also carried out by infecting patients with P. p. with European and African relapsing fever. The results of this therapy are less pronounced, but it is convenient, because the grafting material, previously obtained as a result of infection of the mouse, can be transported over long distances. Somatically weakened patients are vaccinated with Japanese rat typhus - sodoku. In cases where infection fails, as well as in case of somatic contraindications, pyrogenic substances (pyrogenal, etc.) are prescribed.
Since the 40s 20th century carry out complex treatment - pyrotherapy in combination with the administration of antibiotics. In this case, 40,000,000 units of penicillin are prescribed for the course of treatment; Repeated courses of penicillin therapy are carried out at intervals of up to 2 months. iodine control serol. indicators. In some cases, bicillin is used in combination with bismuth preparations. In combination with antibiotic therapy, inf. therapy (malaria vaccination), which promotes the penetration of antibiotics into brain tissue, and in addition, increases the body's defenses. Inf. Therapy is contraindicated in old age, with marasmus, heart failure, aortic aneurysm, kidney disease, and diabetes. In addition, a treatment with penicillin alone has been developed. Several treatment regimens with penicillin have been proposed, but some researchers consider the use of penicillin alone to be insufficient.
Oculomotor disorders in progressive supranuclear palsy
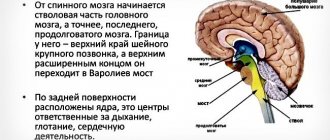
With progressive supranuclear palsy, neurodegeneration in most cases begins in the subcortical and brainstem formations. The cerebral cortex is initially affected to a lesser extent, but as the disease progresses, the process steadily spreads to it. In this case, the anterior parts of the cerebral hemispheres are predominantly affected.
Localization of main changes:
- substantia nigra;
- subthalamic and peduncular nuclei;
- pale globe;
- thalamus;
- midbrain tegmentum;
- stem part of the reticular formation;
- temporal and prefrontal zones of the cerebral cortex.
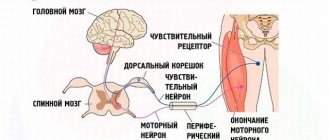
Primary damage to these areas explains the characteristic order of onset of symptoms and the typical Parkinson-like onset of the disease. And the obligatory presence of oculomotor disorders is associated with degeneration of the dorsal part of the midbrain, which leads to a disconnection of connections between the gaze centers in the cortex and brain stem. The nuclei of the cranial nerves themselves, which are responsible for the functioning of the muscles of the eyeballs, remain intact. That's why the paralysis is called supranuclear.
They are a mandatory manifestation of the disease and are characterized by a characteristic combination of symptoms:
- Loss of the ability to move the eyeballs voluntarily: usually first in the horizontal and then in the vertical plane. The outcome is complete ophpalmoplegia with the inability to purposefully move the gaze.
- Decreased convergence, which already in the early stages of the disease is accompanied by complaints of blurred vision and double vision when looking at objects at different distances.
- Preservation of reflex friendly movements of the eyeballs.
- The appearance of the phenomenon of doll's eyes, when the eyeballs, when moving the head, continue to involuntarily fixate the object. This is explained by the lack of suppression of vestibulo-ocular syndrome. At earlier stages, there is intermittency and “lag” in the gaze’s tracking of a moving object (on examination, a neurological hammer), which leads to the appearance of spasmodic “catching up” movements of the eyeballs.
- Gradual decay of the amplitude and speed of voluntary saccadic movements of the eyeballs. During a neurological examination, this is revealed when checking tracking movements; each repeated shift of gaze to the extreme lateral leads is accompanied by an increasing limitation of eye mobility (hypometry).
- Absence of spontaneous nystagmus.
The method of provoking optokinetic nystagmus by rotating the striped drum in front of the patient's face can be used. With progressive supranuclear palsy, the fast phase of nystagmus in the vertical plane initially slows down, and in later stages of the disease it is not caused at all.
The first signs of ophthalmopathy usually appear in the early stages of the disease. Moreover, a decrease in convergence, changes in optokinetic nystagmus and a decrease in voluntary vertical saccades are often detected even when the patient has no complaints of visual impairment. To make a presumptive diagnosis, it is necessary to have at least a limitation of downward gaze, in combination with other signs of a neurodegenerative process.
Symptoms
The clinical picture of such a disease will progress along with the severity of its course. So, at the initial stages of development there are:
- sleep disorders – this includes insomnia or daytime drowsiness;
- causeless irritability;
- chronic fatigue;
- constant weakness;
- severe headaches;
- decreased ability to work - due to the fact that a person cannot fully perform his duties, he is forced to change his job to an easier one;
- frequent mood swings - rudeness and arrogance often come to the fore;
- decreased visual acuity.
At the stage of dementia there are:
- severe memory impairment;
- loss of simple skills;
- inability to recognize loved ones and friends;
- loss of criticality to surrounding events;
- inability to analyze what is happening;
- disorientation;
- lack of initiative;
- increased anxiety;
- personal changes;
- depression;
- psycho-emotional arousal.
At later stages they join:
- clouding of consciousness;
- speech dysfunction;
- loss of ability to count;
- muscle weakness in the upper and lower extremities, as well as in other parts of the body;
- loss of short-term memory;
- delusions and hallucinations;
- capriciousness and short temper;
- seizures;
- changes in pupil size and reactivity;
- weakness and general exhaustion - for this reason a person is forced to constantly stay in bed;
- paresis and paralysis;
- attacks of loss of consciousness.
If such symptoms are ignored, profound dementia develops and the likelihood of death increases significantly.
Diagnosis
Diagnosis in the stage of full development of the disease usually does not cause difficulties. It is established on the basis of psychopathological, neurological manifestations and laboratory data. research. Increasing phenomena of total dementia with a lack of criticism, dysarthria, impaired pupillary reactions (see Argyll Robertson syndrome), persistence of serols.
indicators - all this together makes the diagnosis of P. p. reliable. The Wasserman reaction in the blood is in most cases positive (see Wasserman reaction); in the cerebrospinal fluid, it, like the protein reactions (Nonne-Apelt, Pandi, Weichbrodt), is sharply positive (see Coagulation tests, Cerebrospinal fluid).
The number of cellular elements in the cerebrospinal fluid is increased, sometimes significantly. The total protein content is increased. When performing the Lange reaction (see Cerebrospinal fluid), there is a discoloration of the liquid in the first 4-6 tubes and an increase in color intensity in the next ones (the so-called
Differential diagnosis is carried out with syphilis of the brain (see Syphilis), in which dementia is lacunar in nature with greater or lesser preservation of criticism, hallucinations are more often observed; The Lange reaction curve has the so-called syphilitic tooth. P. p. is differentiated from alcoholic pseudoparalysis (see.
Alcoholic encephalopathy) and senile dementia (see) based on serol. indicators; with brain injuries (see), especially the frontal lobe, according to neurol, symptoms and serol. indicators. Pseudoparalytic syndrome in brain tumors is accompanied by increased intracranial pressure (see.
Hypertensive syndrome). Psychoses of vascular origin differ from P. p. by the development of lacunar dementia (see Atherosclerosis), which is not characteristic of P. p. The presence of paralytic dementia makes it possible to distinguish the circular form of P. p. from manic-depressive psychosis (see), and the hallucinatory-paranoid form - from schizophrenia (see).
Anatomical data
With progressive paralysis, nerve cells in the brain and often the spinal cord die. The arrangement of ganglion cells in rows and layers is disrupted. Glia cells and fibers grow, increase in number and thicken. Already with a superficial examination, it is possible, based on the degree of increase in the number and especially thickening of these elements, to distinguish this proliferation of glia from that which occurs in senile forms. Mitoses are often observed in glial cells. The membranes of small vessels of the cerebral cortex (and other organs) are infiltrated by round cells, mostly plasmatic in nature. The latter are considered characteristic of progressive paralysis, since they are not observed in any other disease except sleepy, which does not occur in our country. New capillary formation can often be clearly seen.
Macroscopically, there is a decrease in the brain; in advanced cases, the weight reaches 1000 grams, the surface is uneven due to the process of wrinkling, the gyri are narrowed, the furrows are widened. The white matter is dirty in color and contracts when cut, unless the atrophy is masked by cerebral edema.
Nowhere is hemorrhagic pachymeningitis as common as in progressive paralysis; the dura mater is often fused to the skull.
Secondary degenerations are also observed in the spinal cord, since there are brain lesions, but in addition, the primary changes that occur in it are often the same as in the brain.
Chronic degeneration is sometimes observed in the peripheral nervous system. The aorta is mostly syphilitically changed. And other organs, especially the liver, do not remain normal. It should be noted that the usual manifestations of syphilis and their consequences (with the exception of vascular phenomena) are rare in paralytics.
Forecast
The duration of the course of untreated P. from its initial manifestations to death is on average approx. 2Y2 years. Youth paralysis progresses more slowly (5-6 years), stationary paralysis - up to 20 years or more. The agitated form ends with the death of the patient after a few months. In the expansive form, long-term remissions are observed.
Prevention consists of timely treatment of syphilis, which is carried out with specific and nonspecific means according to certain schemes (see Syphilis).
Progressive supranuclear palsy is characterized by a steady progression of the neurodegenerative process and an increase in symptoms. The most disabling factor initially is postural disturbances, and in more severe stages the consequences of immobility and swallowing disorders begin to become increasingly important.
The cause of death in progressive supranuclear palsy is usually intercurrent infections, aspiration pneumonia, and sleep apnea.
Causes
The etiological factor of the disease is the causative agent of syphilis - Treponema pallidum. The source of infection is a sick person, the main route of infection is sexual and injection. The pathogen can be transmitted through saliva. The introduction of an infectious agent into cerebral tissue occurs through hematogenous and lymphogenous routes.
The reason for the long-term persistence of the pathogen, which is associated with progressive paralysis, is not known with certainty. Possible predisposing factors are the absence or insufficiency of correct treatment of early forms of syphilis, hereditary predisposition, and alcoholism. Triggers that provoke the activation of infection are considered to be traumatic brain injury, weakened immunity against the background of acute and chronic somatic diseases.
The etiofactors that trigger degenerative processes of a certain cerebral localization remain unknown. Most cases of the disease are sporadic. Individual familial variants with presumed autosomal dominant inheritance have been identified since 1995. Molecular genetic studies have shown that some forms of PSP are caused by defects in the tau protein-encoding gene located at the 17q21.31 locus. The most likely is a multifactorial mechanism for the occurrence of pathology, which is realized against the background of genetic predisposition.
Social and forensic psychiatric significance
Only in isolated cases (persistent remission after treatment) can the patient be allowed to return to his previous professional activity. During a forensic psychiatric examination (see) of the patient, insanity is established in almost all cases (see). Only with treated P. p. with a stable remission lasting at least 3 years can the patient be declared sane.
Bibliography: Venereal diseases, ed. O. K. Shaposhnikova, p. 174, M., 1980; Gordova T. N. Clinic and course of progressive paralysis treated with malaria, M., 1959, bibliogr.; Gurevich M. O. Pathological features of progressive paralysis in connection with clinical data and treatment of malaria, in the book: Psychiat.
hospital on the path to reconstruction, ed. I. N. Koganovich, p. 179, M.-L., 1934; Miniovich P. A. Malarial therapy of neurolesia and other diseases of the nervous system, Rostov n/D., 1934; R o-zenblum A. S. On the relationship of febrile illnesses to psychoses, Works of doctors Odessa. mountains hospitals, in 2, p. 73, Odessa, 1876; Bruetsch W.L.
Neurosyphilitic conditions, Amer. handb. psy-chiat., ed. by S. Arieti, v. 2, p. 1003, NY, 1959, bibliogr.; Gerstmann J. Die Malariabehandlung der progressiven Paralyse, Wien, 1928; Handbuch Geisteskrankheiten, hrsg. v. O. Bumke, Bd 8, T. 4, S. 147, 315, B., 1930, Bibliogr.; W a g-ner-Jauregg J.
D. S. Ozeretskovsky.