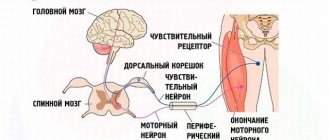
Моторные нейроны — это нервные клетки, которые посылают электрические сигналы на мышцы, влияющие на способность мышц функционировать.
Болезнь моторных нейронов (MND) может появиться в любом возрасте, но большинству пациентов старше 40 лет при диагностике. Она затрагивает мужчин больше, чем женщин.
Наиболее распространенный тип, боковой амиотрофический склероз (БСА), по всей вероятности, поражает около 30 000 американцев в любой момент времени, с более чем 5 000 диагнозов в год.
Известный английский физик Стивен Хокинг прожил с ALS много десятилетий до своей смерти в марте 2020 года. Виртуоз-гитарист Джейсон Беккер — еще один пример того, кто живет с ALS уже несколько лет.
Вот некоторые ключевые моменты, касающиеся заболеваний двигательных нейронов. Более подробно об этом говорится в основной статье.
- Болезни двигательных нейронов (MND) — это группа заболеваний, которые воздействуют на нервные клетки, посылающие сообщения в мозг.
- Происходит постепенное ослабление всех мышц тела, что в конечном итоге сказывается на способности к дыханию.
- Генетические, вирусные и экологические проблемы могут играть определенную роль в возникновении МГП.
- Лекарства не существует, но поддерживающее лечение может улучшить качество жизни.
- Ожидаемая продолжительность жизни после постановки диагноза составляет от 3 лет до более 10 лет.
Поражаемые области
С постепенным развитием заболевания двигательных нейронов у пациентов проявляется нарастание проблем с дыханием, глотанием, движением и общением с другими людьми. Могут проявляться следующие затруднения:
- Кисти, руки. Даже при выполнении ежедневных дел (мытья посуды, поворачивание крана, нажатие клавиш) будут проявляться все большие трудности с усилением слабости в ладонях и руках.
- Ноги. Из-за ослабления ног пациенту будет все труднее передвигаться.
- Эмоциональное состояние. Иногда могут поражаться зоны, которые отвечают за эмоциональные реакции, что проявляется непроизвольным смехом или плачем физиологического характера.
- Плечи, шея. Через определенное время пациенту становится трудно дышать из-за слабеющих мышц шеи.
- Дыхание. В основном заболевание распространяется на дыхательные мышцы, затрудняя дыхание.
- Глотание, речевые функции. Могут быть затруднения с приемом пищи, питьем и разговором.
У некоторых пациентов БДН вызывает нарушения памяти, сниженную концентрацию внимания, сложности с выбором слов и обучаемостью. То есть происходит незаметное и скрытое изменение психических функций.
При БДН остается не нарушенным интеллект, пациенты осознают все происходящее. У многих людей не поражается зрение, вкус, осязание и слух, остаются работоспособными функции мочевого пузыря, кишечника, половых органов (вплоть до поздних стадий) и сердечной мышцы.
диагностирование
На ранней стадии, МГП может быть трудно диагностировать, потому что признаки и симптомы являются общими для других заболеваний, таких как рассеянный склероз (РС), воспаление нервов, или болезнь Паркинсона.
Врач ПМСП обычно направляет пациента к неврологу, врачу, специализирующемуся на диагностике и лечении заболеваний и состояния нервной системы.
Невролог начнет с полного анамнеза и физического обследования неврологической системы.
Могут быть полезны и другие тесты.
Анализ крови и мочи: Эти анализы могут исключить другие условия и выявить любое повышение уровня креатининовой киназы. Он образуется, когда мышцы ломаются, и иногда его можно обнаружить в крови пациентов с МГП.
МРТ-сканирование мозга: Это не может обнаружить МГП, но может помочь исключить другие состояния, такие как инсульт, опухоль мозга, проблемы с циркуляцией мозга или аномальную структуру мозга.
Электромиография (ЭМГ) и исследование нервной проводимости (НЭС): Они часто выполняются вместе. ЭМГ измеряет уровень электрической активности в мышцах, в то время как NCS измеряет скорость, с которой электричество проходит через мышцы.
Спинномозговая пункция или поясничная пункция: При этом анализируется спинномозговая жидкость, жидкость, которая окружает мозг и спинной мозг.
Биопсия мышц: Если врач считает, что у пациента может быть мышечное заболевание, а не МГП, может быть проведена биопсия мышц.
После проведения анализов врач обычно наблюдает за пациентом в течение некоторого времени, прежде чем подтвердить наличие МГП.
Критерии, известные как критерии Эль-Эскориала, могут помочь врачу проверить наличие отличительных неврологических признаков, которые могут помочь в диагностике ALS.
- сокращение мышц, слабость или судороги.
- ригидность мышц или аномальные рефлексы
- симптомы распространяются на новые группы мышц
- не имея других факторов, объясняющих симптомы.
Патогенез и патоморфология болезни
Болезнь мотонейрона считается патологией мультифакторного типа, развитие которой происходит от воздействия генетической наследственности с окружающими факторами.
В 10% ситуаций болезнь связана с семейными формами, где 25% — характеризуются мутированием гена СОД-1. Иногда спорадические виды патологии связаны с изменениями других генов (нейрофиламентов) в результате нарушений функций или структуры, при действии которых мотонейроны начинают дегенерировать. Механизм запускается при действии провокаторов (возраста, пола, тяжелых металлов и пр.). Маловероятной является взаимосвязь появления заболевания от инфекционных агентов.
Клинически нейроны дегенерируют при разрушении от 80% клеточных структур, что затрудняет своевременную терапию патологии и нуждается в диагностических исследованиях во время доклинического этапа.
Возле лобных долей, в 3, 5 слоях центральных извилин, двигательных ядрах 5, 7, 9 и 11 нервов черепа в стволе мозга определяется дегенерация нейронов и изменение кортикоспинальных путей. На различных этапах изменения мотонейронов определяются эозинофильные, базофильные включения и тельца Буниной.
Аксональные сфероиды образуются а проксимальных отделах аксонов. На основании полученных данных, отмечаются нарушения перемещения и деградации белков. В мышечных структурах определяется денервационная атрофия.
Примечания
- Disease Ontology release 2019-05-13 — 2019-05-13 — 2020.
- Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- Epidemiology of Sporadic ALS. (англ.) (недоступная ссылка). Stanford Medicine » School of Medicine. Дата обращения 24 октября 2015. Архивировано 8 октября 2020 года.
- Conwit, Robin A.
Preventing familial ALS: A clinical trial may be feasible but is an efficacy trial warranted? (англ.) // Journal of the Neurological Sciences (англ.)русск. : journal. — 2006. — December (vol. 251, no. 1—2). — P. 1—2. — ISSN 0022-510X. — doi:10.1016/j.jns.2006.07.009. — PMID 17070848. - Al-Chalabi, Ammar; P. Nigel Leigh.
Recent advances in amyotrophic lateral sclerosis (англ.) // Current Opinion in Neurology (англ.)русск.. — Lippincott Williams & Wilkins (англ.)русск., 2000. — August (vol. 13, no. 4). — P. 397—405. — ISSN 1473-6551. — doi:10.1097/00019052-200008000-00006. — PMID 10970056. - «Болезнь Хокинга» объяснили появлением четырехспиральной ДНК // Лента.ру, 2014-03-07
- Cyrille Garnier, François Devred, Deborah Byrne, Rémy Puppo, Andrei Yu. Roman.
Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates (En) // Scientific Reports. — 2017-07-28. — Т. 7, вып. 1. — ISSN 2045-2322. — doi:10.1038/s41598-017-07215-7. - Aphrodite Caragounis, Katherine Ann Price, Cynthia P.W. Soon, Gulay Filiz, Colin L. Masters.
Zinc induces depletion and aggregation of endogenous TDP-43 // Free Radical Biology and Medicine. — Т. 48, вып. 9. — С. 1152—1161. — doi:10.1016/j.freeradbiomed.2010.01.035. - ALSUntangled: исследование болезни Лайма и препарата Iplex = ALSUntangled Update 1: Investigating a bug (Lyme Disease) and a drug (Iplex) on behalf of people with ALS : [пер. с англ.] // ALS INFO : [электр. ресурс]. — 2020. — 25 апреля.
- ALSUntangled Update 1: Investigating a bug (Lyme Disease) and a drug (Iplex) on behalf of people with ALS : [англ.] : Report / The ALSUntangled Group // Amyotrophic Lateral Sclerosis : журн. — 2010. — Vol. 10, no. 4. — P. 248–250. — doi:10.1080/17482960903208599. — PMID 19701824.
- Хондкариан О. А., Боковой амиотрофический склероз, в кн.: Многотомное руководство по неврологии, под ред. С. Н. Давиденкова, т. 3, кн. 1, М., 1962
- Амиотрофический боковой склероз
— статья из Большой советской энциклопедии. - https://www.ninds.nih.gov/disorders/amyotrophiclateralsclerosis/detail_ALS.htm Архивная копия от 4 января 2020 на Wayback Machine «In 90 to 95 percent of all ALS cases, the disease occurs apparently at random with no clearly associated risk factors. … About 5 to 10 percent of all ALS cases are inherited. The familial form of ALS usually results from a pattern of inheritance that requires only one parent to carry the gene responsible for the disease. Mutations in more than a dozen genes have been found to cause familial ALS.»
- Exposure to pesticides and risk of amyotrophic lateral sclerosis: a population-based case-control study.By Bonvicini F, Marcello N, Mandrioli J, Pietrini V, Vinceti M. In Ann Ist Super Sanita. 2010; 46(3):284-7.PMID 20847462
- Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: A meta-analysis of epidemiological studies: Pesticide exposure as a risk factor for ALS. By Malek AM, Barchowsky A, Bowser R, Youk A, Talbott EO. In Environ Res. 2012 Aug; 117:112-9. PMID 22819005
- Are environmental exposures to selenium, heavy metals, and pesticides risk factors for amyotrophic lateral sclerosis?. By Vinceti M, Bottecchi I, Fan A, Finkelstein Y, Mandrioli J. In Rev Environ Health. 2012; 27(1):19-41. PMID 22755265
- Pesticide exposure and amyotrophic lateral sclerosis. By Kamel F, Umbach DM, Bedlack RS, Richards M, Watson M, Alavanja MC, Blair A, Hoppin JA, Schmidt S, Sandler DP. In Neurotoxicology. 2012 Jun; 33(3):457-62. PMID 22521219
- Рефлексогенные зоны
— статья из Большой советской энциклопедии. - Brooks B.R., Miller R.G., Swash M., Munsat T.L.
El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis (англ.) // Amyotroph. Lateral Scler. Other Motor Neuron Disord. : journal. — 2000. — December (vol. 1, no. 5). — P. 293—299. — PMID 11464847. - ↑ 12
https://www.ninds.nih.gov/disorders/amyotrophiclateralsclerosis/detail_ALS.htm Архивная копия от 4 января 2020 на Wayback Machine How is ALS treated? - Эрон Гитлер, Леонард Петручелли.
Разгадка тайны БАС // В мире науки. — 2020. — № 8/9. — С. 50—56. - Неврология. Национальное руководство. — ГЭОТАР-Медиа, 2010. — 2116 с. — 2000 экз. — ISBN 978-5-9704-0665-6.
- Riluzole // PATIENT & CAREGIVER EDUCATION (рус.)
- https://www.ninds.nih.gov/disorders/amyotrophiclateralsclerosis/detail_ALS.htm Архивная копия от 4 января 2020 на Wayback Machine However, the Food and Drug Administration (FDA) approved the first drug treatment for the disease—riluzole (Rilutek)—in 1995. Riluzole is believed to reduce damage to motor neurons by decreasing the release of glutamate. Clinical trials with ALS patients showed that riluzole prolongs survival by several months
- Nagata, Kazuaki
. Japan recognizes Cyberdyne’s robotic suit as medical device, widespread use anticipated (англ.),
The Japan Times Online
(26 November 2015). Дата обращения 10 февраля 2020. - https://mosmedpreparaty.ru/news/4923
- https://www.ab-science.com/file_bdd/content/1490200983_SLAPresentationv03.2017vdef.pdf
- Епископ РОСХВЕ просит главу Минздрава РФ изменить ситуацию с отношением к больным «непризнанным» недугом, NewsRu, 21 августа 2013
- Где взять право на надежду? // «Честное слово» № 3 (832), 23.01.2013
Что является причиной гибели двигательных нейронов?
По мнению специалистов, основными причинами БДН является наследственность и средовые факторы, повышающие риск патологии.
Примерно у 5-10% пациентов с болезнью моторного нейрона была выявлена семейная наследственная форма. В основном БДН была диагностирована (90-95% случаев) при спорадической форме, которая появляется по неизвестным причинам.
По эпидемиологическим исследованиям была выявлена возможная связь действия шоковых, механических травм, высоких спортивных нагрузок, вредных веществ на развитие заболевания.
Сейчас проводятся обследования механизмов повреждения внутриклеточных процессов, из-за которых погибают двигательные нейроны с окружающими клетками.
К возможным причинам относят забастовку и сбой работы «редакторов» в обработке РНК, изменения химических коммуникативных связей спинного мозга, нарушения транспортировки продуктов метаболизма, питательных веществ, скопления агрегатов белка в нейронах с нарушением нормальных функций и склеиванием, появление свободных радикалов кислорода, нарушение энергетического фона клеток и недостаток питательных веществ для мотонейронов. Также гибель двигательных нейронов может возникать из-за окружения глиальными клетками, которые могут быть токсичными.
Причины
Моторные нейроны посылают сигналы от мозга к мышцам и костям, и это заставляет мышцы двигаться. Они участвуют как в сознательных движениях, так и в автоматических движениях, таких как глотание и дыхание.
Некоторые МГП наследуются, в то время как другие происходят случайным образом. Точные причины неясны, но Национальный институт неврологических заболеваний и инсульта (NINDS) отмечает, что генетические, токсичные, вирусные и другие факторы окружающей среды, вероятно, играют определенную роль.
Основные и редкие виды БДН
Заболевание двигательных нейронов имеет несколько основных форм:
- БАС (боковой амиотрофический склероз) развивается у 80% людей с болезнью двигательного нейрона, вызывает в основном слабость, судороги и истощение в мышцах, скованность верхних и нижних конечностей;
- ПБП (бульбарный паралич прогрессирующего типа) поражает около 10-25% людей, распространяется на нейроны спинного и головного мозга, проявляется затруднениями в жевании, глотании, неразборчивости речи;
- мышечная прогрессирующая атрофия (8%) – редкий тип БДН, распространяется на двигательные нейроны в нижних конечностях, развивается слабость, атрофия мышц, фасцикуляции и потеря веса;
- ПЛС (латеральный первичный склероз) (2%) поражает преимущественно двигательные нейроны в верхних конечностях, развивая спастику мышц, неустойчивую ходьбу, затрудненную речь.
Симптомы болезней мотонейронов
Ниже приводится краткое описание симптомов некоторых из наиболее распространенных форм болезней двигательных нейронов.
Боковой амиотрофический склероз (БАС), также называемый болезнью Лу Герига или классической болезнью моторных нейронов, является прогрессирующим, в конечном счете имеющим смертельный исход заболеванием, которое нарушает передачу сигналов во всех мышцах организма. Многие врачи используют термины «болезнь моторных нейронов» и БАС как взаимозаменяемые. Причиной болезни являются нарушения как верхних, так и нижних двигательных нейронов. Первые симптомы обычно отмечаются в руках и ногах или в мышцах, отвечающих за глотание. Примерно у 75 процентов больных классическим БАС развиваются слабость бульбарных мышц (мышцы, которые контролируют речь, глотание и жевание). Мышечная слабость и атрофия происходят по обе стороны тела. Больные теряют силу и способность двигать руками и ногами и удерживать тело в вертикальном положении. Другие симптомы включают в себя спастичность мышц, спазмы, мышечные судороги. Речь может стать невнятной или гортанной. Когда мышцы диафрагмы и грудной стенки не функционируют должным образом, больные теряют способность дышать без механической поддержки. Хотя болезнь как правило не нарушает интеллектуальные способности человека или личности, отдельные недавние научные исследования показывают, что у некоторые больных БАС могут развиваться когнитивные (умственные) нарушения. Большинство людей, страдающих боковым амиотрофическим склерозом, умирают от респираторной недостаточности, обычно через 3 — 5 лет с момента появления симптомов. Однако около 10 процентов больных выживают в течение 10 или более лет.
Прогрессивный бульбарный паралич, также называемый прогрессирующей атрофией бульбарной группы черепно-мозговых нервов, воздействует на нижние двигательные нейроны, отвечающие за такие действия, как глотание, речь, жевание и другие. Симптомы включают в себя слабость языкоглоточной мышцы, челюстной и лицевых мышц, прогрессирующую потерю функции речи и атрофию мышц языка. Слабость конечностей при заболевании с признаками повреждения двигательного нейрона почти всегда очевидна, но менее заметна. Люди подвержены повышенному риску удушья и аспирационной пневмонии, вызванной прохождением жидкостей и пищи через нижние дыхательные пути и легкие. У пострадавших присутствуют эмоциональные вспышки смеха или плача (называемые эмоциональной лабильностью). Инсульт и миастения могут иметь определенные симптомы, сходные с симптомами прогрессирующего бульбарного паралича и должны быть исключены при диагностике этого заболевания. Примерно у 25 процентов людей с БАС ранние симптомы начинаются с сопутствующим участием бульбарных нарушений. Многие клинические врачи считают, что прогрессирующий бульбарный паралич сам по себе, без признаков патологий в конечностях (руках или ногах), встречается крайне редко.
Псевдобульбарный паралич, имеющие многие симптомы, аналогичные прогрессирующему бульбарному параличу, характеризуется дегенерацией верхних моторных нейронов, которые передают сигналы нижним моторным нейронам в стволе мозга. У больных развивается прогрессирующая потеря способности говорить, жевать и глотать, а также прогрессивная слабость лицевых мышц. У больных могут развиться голосовые нарушения и повышенный рефлекс кляпа. Язык может стать неподвижным и потерять способность выступать из рта.
Первичный боковой склероз (ПБС) повреждает верхние двигательные нейроны рук, ног и лица. Это происходит, когда специфические нервные клетки в моторных областях коры головного мозга (тонкий слой клеток, покрывающих мозг, который отвечает за большинство функций мозга на высоком уровне) постепенно вырождаются, заставляя движения быть медленными. Болезнь часто затрагивает сначала ноги, а затем туловище, руки и, наконец, бульбарные мускулы. Речь может становится замедленной и вызывать сложности. При поражении нервных клеток движения ног и рук становятся неуклюжими, медленными и слабыми, возникает спастичность, что приводит к невозможности ходить или выполнять задачи, требующие точной координации рук. Проблемы с равновесием могут привести к падению. Речь может стать медленной и невнятной. Больные обычно испытывают псевдобульбарный аффект и чрезмерно активный ответ. Первичный боковой склероз чаще встречается у мужчин, чем у женщин, начало болезни обычно происходит между 40 и 60 годами. Причина заболевания неизвестна. Симптомы прогрессируют постепенно в течение многих лет, что приводит к прогрессивной жесткости (спастичности) и неуклюжести пораженных мышц. ПБС иногда считается формой бокового амиотрофического склероза, но основное отличие заключается в сохранении более низких моторных нейронов, медленной скорости прогрессирования заболевания и нормальной продолжительности жизни. Первичный боковой склероз может ошибочно приниматься за спастические параплегии, наследственное расстройство верхних моторных нейронов, которое вызывает спазм в ногах и как правило начинается в подростковом возрасте. Большинство неврологов следуют клиническому течению пострадавшего человека в течение как минимум 3-4 лет, прежде чем поставить диагноз. Данное заболевание не смертельно, но может оказывать влияние на качество жизни.
Прогрессивная мышечная атрофия характеризуется медленной, но прогрессирующей дегенерацией исключительно нижних моторных нейронов. Заболеванию подверждены в значительной степени мужчины, с началом заболевания раньше, чем при других болезнях мотонейронов. Слабость обычно возникает сначала в руках, а затем распространяется в нижнюю часть тела, где она может быть в более тяжелой форме. Другие симптомы могут включать в себя мышечное истощение, неуклюжие движения рук и мышечные судороги. Могут пострадать мышцы, отвечающие за дыхание. Воздействие холода может ухудшить симптомы. Болезнь развивается совместно с БАС во многих случаях.
Спинальная мышечная атрофия (СМА) является наследственной болезнью, воздействующей на нижние двигательные нейроны. Это аутосомно-рецессивное заболевание, причиной которого являются нарушения в гене SMN1 (ген ответственный за производтсво белка, который важен для функционирования моторных нейронов (SMN-белок)). При СМА недостаточный уровень белка SMN приводят к дегенерации нижних моторных нейронов, вызывая слабость и истощение скелетных мышц. Слабость часто более выражена в мышцах рук и ног, а также в мышцах туловища. Спинальная мышечная атрофия у детей подразделяется на три типа, исходя из возраста начала, тяжести и прогрессирования симптомов. Все три типа вызваны дефектами в гене SMN1.
Синдром постполиомиелита (СПП) — это состояние, которое может поражать больных, перенесших полиомиелит, что может произойти в течение десятилетий после их выздоровления от полиомиелита. Полиомиелит — острое вирусное заболевание, которое разрушает моторные нейроны. Многие люди, пострадавшие от заболевания на ранней стадии жизни, восстанавливаются, а новые симптомы возникают спустя много десятилетий. После острого полиомиелита выжившие двигательные нейроны отвечают за большее количество мышц, которые они контролируют. Считается, что синдром постполиомиелита и постполиотическая мышечная атрофия возникают, когда выжившие моторные нейроны утрачиваются в процессе старения или вследствие травмы/болезни. Многие ученые считают, что СПП является скрытым симптомом слабости мышц, ранее пораженных полиомиелитом, а не новой болезнью моторных нейронов. К симптомам относится усталость, медленно прогрессирующая мышечная слабость, мышечная атрофия, непереносимость холода, боль в мышцах и суставах. Эти симптомы чаще всего проявляются среди групп мышц, пораженных начальным заболеванием, и могут состоять из проблем при дыхании, глотании или во время сна. Другие симптомы СПП могут быть вызваны скелетными деформациями, такими как давно сформировавшийся сколиоз, которые привели к хроническим изменениям в биомеханике суставов и позвоночника. Симптомы чаще встречаются у пожилых людей и тех лиц, которые в наибольшей степени страдают от более раннего заболевания. У некоторых людей наблюдаются только незначительные симптомы, в то время как у других развивается мышечная атрофия, которую можно ошибочно диагностировать как БАС. СПП как правило не угрожает жизни больного. По медицинской статистике, у 25 — 50 процентов перенесших паралитический полиомиелит обычно развивается синдром постполиомиелита.
Симптоматика и клиника БДН в зависимости от формы
На моторные нейроны влияет целая группа болезней. В состав нейронов входят две группы клеток, необходимые для произвольных движений.
В первую группу входят двигательные нейроны, расположенные в прецентральной мозговой извилине, а во вторую – нейроны, расположенные в стволе спинного и головного мозга.
В зависимости от локализации погибшего двигательного нейрона будут зависеть форма болезни и ее симптомы.
При спастической параплегии поражается двигательный нейрон первой группы, при спинальной мышечной атрофии, детском параличе – вторая группа, а при боковом амиотрофическом склерозе обе группы нейронов.
Формы БДН и их проявления:
- Спастическая параплегия определяется повышением спастического тонуса ног с нарушением походки. Это вызывается генетическими мутациями.
- Для спинальной мышечной атрофии характерна слабость и уменьшение мышц, отсутствие их активации или иннервирования нервными волокнами. В зависимости от времени появления первой симптоматики и характеру распределения слабости мышц, осуществляется классификация спинальной мышечной атрофии.
- Более распространенной патологией опорно-двигательной системы является БАС, частота развития которого повышается с возрастом. В результате прогрессирует омертвление нервных клеток, нарастает слабость и мышечная атрофия.
- Детский церебральный паралич, полиомиелит является вирусной болезнью нижних нейронов.
Диагностика болезни моторных нейронов
Не существует каких-либо специальных исследований для диагностики большинства болезней мотонейронов, хотя в настоящее время существуют генетическое тестирование для генов SMA. Симптомы могут различаться у больных и на ранних стадиях заболевания могут быть похожими на симптомы других заболеваний, что затрудняет диагностику. За физическим осмотром следует тщательное неврологическое обследование. Неврологические тесты будут оценивать двигательные и сенсорные навыки, функционирование нервной системы, слух и речь, зрение, координацию и равновесие, психическое состояние и изменения настроения или поведения.
Исследования для исключения других заболеваний или измерения степени поражения мышц, могут включать следующие:
Электромиография (ЭМГ) используется для диагностики расстройств нижних моторных нейронов, а также нарушений мышц и периферических нервов. При ЭМГ врачом вводится тонкий игольчатый электрод, прикрепленный к регистрирующему инструменту, в мышцу для оценки электрической активности во время произвольного сокращения и в состоянии покоя. Электрическая активность в мышце вызвана более нижними двигательными нейронами. Когда двигательные нейроны утрачиваются, в мышце возникают характерные аномальные электрические сигналы. Тестирование обычно длится около часа или более, в зависимости от количества тестируемых мышц и нервов.
ЭМГ как правило проводится в сочетании с исследованием скорости нервной проводимости. Исследования нервной проводимости измеряет скорости передачи импульсов в нервах от небольших электродов, прикрепленных к коже, а также их силу. Небольшой импульс электричества (подобный толчке от статического электричества) стимулирует нерв, который отвечает за конкретную мышцу. Второй комплект электродов передает ответный электрический сигнал на записывающее устройство. Исследования нервной проводимости помогают дифференцировать болезни нижних моторных нейронов от периферической невропатии и могут обнаруживать нарушения в сенсорных нервах.
Лабораторные анализы крови, мочи и других веществ позволяют исключить мышечные заболевания и другие расстройства, которые могут иметь симптомы, сходные с симптомами болезни мотонейронов. Например, анализ спинномозговой жидкости, которая окружает мозг и спинной мозг, может обнаруживать инфекции или воспаление, которые также могут вызывать мышечную жесткость. Анализы крови могут назначаться для измерения уровней белковой креатинкиназы (необходима для химических реакций, которые вырабатывают энергию для мышечных сокращений); высокие уровни позволяют диагностировать мышечные заболевания, такие как мышечная дистрофия.
Магнитно-резонансная томография (МРТ) использует мощное магнитное поле для получения детальных изображений тканей, органов, костей, нервов и других структур тела. МРТ часто используется для исключения болезни, которые воздействуют на голову, шею и спинной мозг. МРТ-изображения позволяют диагностировать опухоли головного и спинного мозга, заболевания глаз, воспаление, инфекцию и сосудистые нарушения, которые могут привести к инсульту. МРТ также может обнаруживать и контролировать воспалительные заболевания, такие как рассеянный склероз. Магнитно-резонансная спектроскопия — это тип МРТ-сканирования, который измеряет уровень химических веществ в головном мозге и может использоваться для оценки целостности верхних моторных нейронов.
Биопсия мышц или нервов позволяет подтвердить наличие неврологических нарушений, в частности нарушения регенерации нервов. Небольшой образец мышечной или нервной ткани берется под местным анестетиком и исследуется под микроскопом. Образец может быть удален хирургическим путем через разрез на коже или путем биопсии, при которой тонкая полая игла вводится через кожу в мышцу. Небольшая часть мышечной ткани остается в полой игле, когда она удаляется из тела. Хотя это исследование может предоставить ценную информацию о степени повреждения, это инвазивная процедура, и многие эксперты считают, что биопсия не всегда необходима для диагностики.
Транскраниальная магнитная стимуляция была впервые разработана как диагностический инструмент для изучения областей головного мозга, связанных с двигательной активностью. Она также используется в качестве лечения некоторых заболеваний. Эта неинвазивная процедура создает магнитный импульс внутри мозга, который вызывает двигательную активность в области тела. Электроды, прикрепленные к различным областям тела, собирают и регистрируют электрическую активность в мышцах. Меры вызванной активности могут диагностировать двигательную нервную дисфункцию при болезни мотонейронов или в мониторинге прогрессирования заболевания.
Дифференциальная диагностика
Диагноз БДН подтверждается элеткронейромиографией, которая выявляет генерализованный денервационный процесс. Для выявления спонтанности, продолжительности двигательных единиц, фасцикуляций, фибрилляций, выполняется трехуровневая игольчатая ЭМГ.
Стимуляционная электронейромиография позволяет выявлять замедление двигательных волокон, пирамидную недостаточность.
Транскраниальная магнитная стимуляция выявляет уменьшение возбудимости в центральных мотонейронах. МРТ позволяет исключать остальные патологии с похожей клиникой (компрессию грыжи или опухолью).
Возможности лечения
Болезнь двигательного нейрона неизлечима, а целью терапии является замедление развития процесса, сокращение выраженности симптоматики, так как не существует методов для полного излечения. Доказана эффективность Рилутека – пресинаптического ингибитора.
Как и при БАС, могут применяться антиоксидантные средства. Также специалисты назначают креатин, карнитин, витамины. Другим лекарственным средством является Рилузол, но средство официально недоступно для пациентов в РФ. Как и Рилутек, препарат снижает объем глутамата, освобождающегося при передаче нервных импульсов.
К полиативной терапии относится устранение болей от мышечных спазмов, для чего назначается Карбамазепин в дозе около 100-200 мг несколько раз в сутки. При увеличенном тонусе мышц нужно принимать миорелаксанты (Сирдалуд, Баклофен).
Дыхательная поддержка – это основной фактор, влияющий на качество жизни и прогноз. С прогрессированием патологии наблюдается ослабление и атрофия мышц диафрагмы и других мышечных групп, требуется неинвазивное вентилирование легких.
Это нормализует сон, снижает утомляемость и одышку. Используется аппарат со специальной маской.
Если нарушены функции глотания, то кормление проводится назогастральным зондом, установкой гастростомы. Назначается специальное питание, восполняющее недостаток калорий.
При заболевании БДН трудно говорить про прогноз. В редких случаях люди живут десятки лет. Человек может умереть через несколько месяцев. Но в среднем длительность жизни равна 2-5 года от начала выявления болезни. Не существует специфических профилактических мер.
Лечение[ | ]
Больным БАС требуется поддерживающая терапия для облегчения симптомов[20].
Постепенно у больных начинает ослабляться дыхательная мускулатура, развивается дыхательная недостаточность и становится необходимым применение оборудования для облегчения дыхания во время сна (IPPV или BIPAP). Затем, после полного отказа дыхательной мускулатуры, требуется круглосуточное использование аппарата искусственной вентиляции лёгких.[20]
Ведутся исследования по методу лечения, использующего блокировку генов, вызывающих это заболевание[21].
Замедление прогрессирования[ | ]
Рилузол (рилутек) — единственный препарат, достоверно замедляющий прогрессирование БАС[22][23]. Доступен с 1995 года. Он ингибирует высвобождение глутамата, тем самым уменьшая повреждение двигательных нейронов. Продлевает жизнь больных в среднем на месяц, немного отдаляет момент, когда больному потребуется искусственная вентиляция легких[24].
HAL-терапия, новый метод роботизированного лечения, официально допущен к применению в реабилитации БАС в Европе и Японии[25].
В 2020 году для лечения БАС впервые за 22 года был одобрен новый препарат — Радикава (Эдаравон)[26].
Также проводятся клинические исследования по применению препарата Маситиниб[27].
В России[ | ]
В Москве действуют:
- Благотворительный фонд помощи людям с БАС и другими нейромышечными заболеваниями «Живи сейчас»
- Благотворительный фонд помощи больным БАС Г. Н. Левицкого. https://www.alsportal.ru
- служба помощи больным БАС при АНО «Больница Святителя Алексия».
В то же время в России многим больным БАС не оказывается надлежащая медицинская помощь[28]. Например, до 2011 года БАС даже не был включен в список редких заболеваний, а единственный препарат, замедляющий течение болезни, Рилузол, не зарегистрирован[29].
БАС — двигательные нейроны отмирают полностью
Во время БАС сочетается равномерно выраженная симптоматика центрального и периферического паралича, преобладание одних симптомов над другими (сегментарно-ядерный или пирамидный варианты). На поздних этапах болезни преобладают показатели периферического паралича.
Во время БАС начинается повреждение мотонейронов в передних рогах спинового мозга. С прогрессированием патологии проявляются бульбарные нарушения в основном у мужчин.
Выделяется грудной, шейный, диффузный и поясничный дебют. Классическим вариантом является шейный, первая симптоматика которого характеризуется слабостью и атрофией кисти, нарушенными мелкими движениями, наличием фасцикулярных подергиваний пораженных мышц.
Стивен Хокинг — самый известный пациент с БАС
В дальнейшем появляется слабость и атрофия мышц плечевого пояса, пациентам сложно одеваться, поднимать руки выше горизонтальной плоскости. Сначала поражается одна сторона тела, затем симптомы проявляются на другой руке с развитием верхнего смешанного парапареза.
Развивается слабость и скованность нижних конечностей, больным сложно ходить на большие расстояния, перемещаться по лестнице.
Повреждаются разгибательные мышцы с проявлением смешанного нижнего парапареза. На поздних стадиях проявляются псевдобульбарный и бульбарный синдромы, появление дисфагии.
Первой симптоматикой грудного дебюта будут фасцикуляции, атрофия мышц в животе, спине и слабость. Пациентам становится сложно стоять и нагибаться. На следующих этапах появляется смешанный односторонний гемипарез с вовлечением мускулатуры противоположной части тела. На начальном этапе выпадают брюшные рефлексы. В результате дыхательных нарушений наступает летальный исход.
Диффузный дебют проявляется признаками нарушения периферических мотонейронов с резко выраженным утомлением, дыхательными нарушениями. Снижается масса тела до появления дисфагии, в результате дыхательной недостаточности наступает смерть.
Обычно поражение жевательных и мимических мышц наступает позже, пациенту сложно высовывать язык, вытягивать в трубочку губы, надувать щеки.
К осложнениям БАС относят параличи, парезы шейных мышц и конечностей, нарушенное глотание, дыхательную недостаточность, контрактуры конечностей, аспирационную пневмонию, уросепсис, депрессию, болезненные спазмы мышц, кахексию. С прогрессированием двигательных нарушений пациент умирает.
Факторы риска
Вот некоторые из факторов риска, связанных с МГП.
Наследственность: В Соединенных Штатах Америки (США) из каждых 10 случаев АЛС наследуется около 1 случая. Также известно, что SMA является наследственным состоянием.
Возраст: после 40 лет риск значительно возрастает, хотя он все еще очень мал. Скорее всего, АЛС возникает в возрасте от 55 до 75 лет.
Секс У мужчин больше шансов развиться МГП.
Некоторые эксперты связывают военный опыт с более высокой вероятностью развития этого заболевания.
Исследования показали, что профессиональные футболисты чаще умирают от АЛС, болезни Альцгеймера и других нейродегенеративных заболеваний, чем другие люди. Это подразумевает возможную связь с повторяющимися травмами головы и неврологическими заболеваниями.