Причины болезни Галлервордена-Шпатца, факторы риска
БГШ является генетическим заболеванием, которое вызывается унаследованным дефектом в гене пантотенат киназы-2 (PANK2). Белок PANK2 контролирует образование в теле коэнзима А. Это соединение помогает организму конвертировать жиры, некоторые аминокислоты и углеводы в энергию.
В некоторых случаях болезнь Галлервордена-Шпатца может быть не связана с мутациями в гене PANK2. Медики полагают, что недуг вызывается дефектом в нескольких других генах. Однако идентифицировать эти гены специалистам пока не удалось.
https://www.youtube.com/watch?v=ytpressru
Факторы риска
Основным фактором риска является наличие в семье родственников с данным расстройством. БГШ обычно проявляется впервые в детском возрасте, до 10 лет. Эта форма заболевания считается ранней. Поздняя форма характеризуется отсутствием симптомов в детском возрасте, но их проявлением после 20-летнего возраста.
Болезнь Галлервордена-Шпатца — это генетическая патология, которая носит как семейный, так и спорадический характер. Заболевание передаются по наследству аутосомно-рецессивным путем. Заболевание возникает на фоне аберраций в гене пантотенаткиназы. Всего выявлено более 50 мутаций. На фоне генетического дефекта отмечается уменьшение продукции пантотенаткиназы, что вызывает накопление в базальных структурах цистеина, который формирует устойчивые химические соединения с ионами железа, которые оказывают негативное воздействие на белки и запускают процесс перекисного окисления, что вызывает гибель нейронов. На месте некротизированных нейронов отмечается разрастание глиальной ткани.
Такие патологические процессы чаще всего формируются в области бледного шара и черного вещества, где морфологически обнаруживаются внеклеточные отложения железа, имеющие коричневую пигментацию. Помимо этого, имеют место сфероидные периаксональные образования, находящиеся в белом церебральном веществе, коре мозга, спинном мозге и периферических нервных стволах.
Данный синдром связан с мутацией в гене PANK2, которая передается по наследству. Механизм передачи — аутосомно-рецессивный. Это значит, что мутантный ген наследуется не связанно с полом и проявится лишь при наличии двух копий. Так, есть два сценария, когда ребенок родится с болезнью:
- Один из родителей болен, а другой — носитель.
- Оба родителя — носители мутантного гена, при этом никаких клинических признаков не имеют.
Ген PANK2 отвечает за производство фермента пантотенаткиназы 2, который участвует в синтезе витамина B5 и коэнзима А. В результате нарушения синтеза фермента при болезни Галлервордена-Шпатца происходит накопление цистеина в базальных ганглиях, а цистеин связывается с ионами железа. Таким образом накапливается железо в веществе головного мозга.
Причины возникновения мутации неизвестны. Определяющим фактором является передача мутантного гена от родителей ребенку. Наличие одной копии гена (носительство) никак не проявляется клинически, поэтому предугадать рождение детей с патологией практически невозможно.
Печально, но не безнадежно
При болезни Галлервордена-Шпатца симптомы постоянно прогрессируют. Наиболее тяжело протекает детская форма заболевания. Полная инвалидизация индивида наступает через 10-15 лет с момента первого проявления клинических проявлений.
Наиболее благоприятное развитие болезни прогнозируется при взрослой форме болезни. Особенно в случае слабой выраженности деменции.
Терапия позволяет сохранить относительное качество жизни пациента и его способность к самообслуживанию. Продолжительность жизни при атипичной форме болезни Галлервордена-Шпатца может составлять более 20 лет.
Наследственная паллидарная дегенерация Галлервордена—Шпатца (болезнь Галлервордена—Шпатца) — редкое наследственное дегенеративное заболевание нервной системы, впервые описанное в 1922 г. К настоящему времени в литературе имеется около 50 детальных описаний семейных случаев болезни Галлервордена—Шпатца (БГШ), опубликован также ряд наблюдений спорадических случаев заболевания.
Болезнь Галлервордена—Шпатца наследуется по аутосомно-рецессивному типу. Ген, ответственный за возникновение болезни, локализуется на коротком плече 20-й хромосомы в локусе 20р12.3-р13. Первичный молекулярный дефект не установлен.
Патологоанатомически болезнь Галлервордена—Шпатца характеризуется двумя основными признаками: 1) отложением железосодержащего пигмента в области бледного шара, ретикулярной части черной субстанции и красных ядер, что придает им характерное желтовато-коричневое окрашивание; 2) наличием в базальных ганглиях, коре больших полушарий, периферических нервах особых сфероидных нейроаксональных образований, представляющих собой локальные расширения аксонов с пролиферацией мембранных и тубулярных структур.
Симптомы болезни Галлервордена-Шпатца, диагностика
Классическим вариантом болезни Галлервордена-Шпатца считается ранняя детская форма с клинической манифестацией в возрасте от 4 до 10 лет. В большинстве случаев заболевание дебютирует с развития торсионной дистонии, которая затрагивает мышцы ног. Основной жалобой является затруднение ходьбы. Позже, происходит генерализация процесса с его распространением на мышцы глотки, лица, туловища.
Наряду с генерализованными вариантами могут отмечаться мультифокальные или сегментарный тип дистонии. Чаще всего у больных возникает блефароспазм, лицевой параспазм и спастическая кривошея. У трети больных выявляются признаки паркинсонизма, обусловленные мышечной ригидностью и гипокинезией. Иногда отмечается развитие эпилептических приступов.
Болезнь Галлервордена-Шпатца сопровождается когнитивными расстройствами, обусловленными снижением внимательности и памяти с постепенным развитием олигофрении, а также психическими расстройствами с преобладанием агрессивности и асоциального поведения. У больных отмечается дизартрия и нарушение остроты зрения.
Подростковый вариант болезни Галлервордена-Шпатца проявляется в возрасте от 10 до 18 лет и обусловлен более замедленным течением. Манифестация недуга начинается с возникновения фокальной торсионной дистонии, наиболее часто в мышцах конечностей или ортомандибулярной области. Недуг арактеризуется развитием психических, интеллектуальных и поведенческих расстройств.
Болезнь Галлервордена-Шпатца вызывает широкий спектр симптомов, которые варьируются в зависимости от тяжести заболевания и степени его прогрессирования.
Общий симптом — искажающие мимику и положение тела мышечные сокращения. Таким сокращениям могут быть подвержены:
- лицо;
- туловище;
- конечности.
Другие симптомы БГШ это:
- атаксия;
- судороги;
- скрюченные, спазмированные, жесткие конечности;
- отсутствие баланса;
- дезориентация в пространстве;
- бред;
- непоследовательность мыслей и поступков;
- ступор;
- слабоумие;
- замедленность жестов;
- недержание мочи;
- слюнотечение;
- затрудненное глотание;
- гримасы, нарушение нормального выражения лица;
- ригидность мышц;
- невозможность сформировать членораздельное предложение;
- болезненность при мышечных спазмах;
- неправильное положение тела.
Наиболее эффективными методами диагностики болезни Галлервордена-Шпатца являются:
- МРТ;
- физический осмотр;
- клиническая картина заболевания;
- генетическое тестирование (возможно не в любой клинике из-за сложности);
- семейная история болезни.
Так как БГШ передается по наследству, первые проявления могут быть уже в детстве. Однако случается, что впервые симптомы появляются после 18 лет.
На этом основании выделяют 3 варианта течения болезни:
- Ранний. Манифестация в возрасте до 10 лет. При этой форме заболевание быстро прогрессирует. Симптомы ярко выражены. Нарушения мышечного тонуса затрудняют ходьбу, речь, глотание. Дети быстро теряют возможность передвигаться самостоятельно.
- Подростковый. Менее злокачественная форма, которая начинается в промежутке от 10 до 18 лет. Признаки заболевания чаще локальны. Прогрессирует медленнее.
- Поздний. Дебют после 18 лет — наиболее благоприятный вариант течения. Чаще всего выражается нарушениями речи. Со временем клиника прогрессирует: развивается паркинсонизм, усугубляются нарушения психики и интеллекта.
Все формы заболевания имеют общие признаки. Каких-то специфических симптомов нет.
https://www.youtube.com/watch?v=ytaboutru
Ведущий синдром — паркинсонизм. Это целая группа неврологических нарушений. Паркинсонизм не тождественен болезни Паркинсона.
- Дистония. Один из ведущих симптомов. Мышечная дистония проявляется насильственными непроизвольными движениями в конечностях. Со стороны постороннего наблюдателя кажется, что пациент бьется в конвульсиях. Значительно нарушается ходьба. Дистония может быть фокальной:
- В конечностях. Проявляется дистоническими позами. Характерна для начала ранней формы заболевания и для других форм при их прогрессировании. При значительных ранних нарушениях ходьбы пациенты быстро теряют способность передвигаться самостоятельно.
- Оромандибулярной. Мышечная дистония в области рта и нижней челюсти. У пациентов наблюдается спазм мышц челюсти, который затрудняет произвольное открывание рта. При этом отмечаются непроизвольные движения: высовывание языка, вытягивание губ. Может быть нарушение глотания, наклон головы, повороты корпуса.
- Потеря центрального зрения, преобладание периферического — больные смотрят на объекты искоса.
- Потеря периферического зрения, преобладание центрального (туннельное зрение).
- Ночная слепота.
- Снижение остроты зрения.
- Неприязнь к ослепляющему яркому свету.
- В итоге — полная потеря зрения.
Особенности клинической картины
Признаки данной патологии могут варьироваться в каждом конкретном случае, и проявление симптомов зависит от формы болезни у индивида.
Детская форма Галлервордена Шпатца проявляется в возрасте между 5 и 10 годами. Считается классической формой болезни.
В подавляющем количестве случаев (90%) заболевание начинается с торсионной дистонии, которая поражает мышцы ног. У больного происходит сокращение мышц, затрудняется ходьба, изменяется походка. Затем поражаются мышцы лица, глотки и туловища.
Возможно присутствие блефароспазма, спазма кистей, спастической кривошеи, лицевого гемиспазма. Треть пациентов имеют мышечную ригидность, гипокинезию, тремор (признаки синдрома паркинсонизма).
К типичным признакам детской формы болезни также относят:
- эпилептический синдром;
- агрессивность;
- умственную отсталость (возникает из-за ухудшения памяти и внимания);
- асоциальность;
- зрительные нарушения (атрофия зрительных нервов, дегенерация сетчатки);
- психические расстройства.
Дифференциальная диагностика
БГШ по клинической картине напоминает многие другие заболевания нервной системы, основным проявлением которых является паркинсонизм.
- Болезнь Вильсона-Коновалова. Патология, связанная с нарушением обмена меди. Передается наследственно по такому же механизму, как и БГШ. Симптомы схожи: дистония мышц, деменция, дизартрия, дисфагия. Есть патогномичный симптом — необычный цвет глаз за счет колец Кайзера-Флейшера. Различают заболевания по результатам МРТ, анализа ДНК.
- Болезнь Гентингтона. Наследственное заболевание нервной системы, которое связано с атрофией стриатума (одна из структур базальных ядер) и коры головного мозга. Проявляется беспорядочными движениями и снижением когнитивных способностей. В отличие от БГШ, чаще появляется после 35 лет. Диагноз выставляется на основании генетического тестирования и МРТ.
- Болезнь Фара. Дегенеративное заболевание неясного происхождения. Как и БГШ, затрагивает базальные ганглии, только вместо железа в них происходит отложение кальция. На МРТ и КТ — симметричное поражение с кальцификатами. Клиническое проявление — паркинсонизм. Впервые симптомы могут появиться в любом возрасте.
Диагностические критерии и методики
Основные критерии для диагностики:
- начало болезни до 30 лет;
- типичная картина в результатах МРТ;
- постоянное развитие симптомов;
- экстрапирамидальные синдромы;
- пирамидные знаки;
- эпилептические приступы;
- когнитивные нарушения;
- пигментная дегенерация сетчатки;
- наличие болезни в семейном анамнезе.
Болезнь необходимо дифференцировать с другими заболеваниями, которые имеют сходную клиническую картину: хореей Гентингтона, болезнью Вильсона-Коновалова, хореоакантоцитозом, болезнью Мачадо-Джозефа.
Для диагностирования пациент направляют на такие обследования:
Лечение болезни Галлервордена-Шпатца
В настоящее время не разработано эффективных методов лечения болезни Галлервордена-Шпатца. В связи с этим применяется симптоматическое лечение. Синдром паркинсонизма служит показанием к назначению дофаминовых агонистов или производных амантадина. Однако при данном заболевании он, как правило, резистентен к проводимому лечению.
При гиперкинезах показан прием вальпроатов, бензодиазепинов. При спастических симптомах рекомендованы миорелаксанты, при эпиприступах — топирамат или вальпроаты, при когнитивных расстройствах — ипидакрин и холина альфосцерат, при психических отклонениях — нейролептики.
Лечение пациентов с болезнью Галлервордена-Шпатца остается в основном симптоматическим, то есть направленным на снятие симптомов, а не на устранение причины. Устранить генетическую аномалию, приводящую к БГШ, в настоящее время не представляется возможным.
Тремор эффективно устраняется дофаминергическими агентами. Антихолинергический препарат бензотропин может быть использован для смягчения ригидности мышц и уменьшения тремора, наравне с агонистами дофамина.
Для устранения дизартрии и слюнотечения рекомендованы:
- бромид метскополамина;
- логопедические манипуляции.
Системные хелатирующие агенты, такие как десферриоксамин, ранее использовались для удаления избытка железа из головного мозга, однако их эффективность не была доказана. Деменция является прогрессирующим симптомом, её прогрессирование можно замедлить, однако вылечить это состояние нельзя.
Рекомендуемые препараты при болезни Галлервордена-Шпатца:
- баклофен;
- бензтропин;
- мемантин;
- ривастигмин;
- донепезил.
Рекомендуемые занятия:
- физиотерапия;
- трудотерапия;
- логопедия.
В данный момент болезнь Галлервордена-Шпатца не поддается лечению. Раньше пытались подействовать на накопление железа клетками базальных ядер. Однако такая схема лечения не оказалась эффективной.
Ведутся исследования препаратов, относящихся к группе детоксицирующих средств. Предполагается, что они способны замедлить течение БГШ. Клинические испытания показали, что это действительно так, однако разница несущественна.
Для облегчения состояния пациента применяют симптоматические средства.
- Лечение паркинсонизма. Дофаминовые агонисты сглаживают симптомы, однако при БГШ они значительно теряют эффективность. Поэтому чаще при гиперкинезах назначают транквилизаторы.
- При мышечной дистонии применяют миорелаксанты.
- При эпилептических приступах — противоэпилептические препараты группы вальпроатов.
- При признаках тяжелых нарушений психической сферы — антидепрессанты и нейролептики.
Терапия помогает улучшить течение заболевания и сгладить основные симптомы. При этом прием лекарственных препаратов никак не влияет на продолжительность жизни и не улучшает прогноз.
https://www.youtube.com/watch?v=https:accounts.google.comServiceLogin
Разрабатываются экспериментальные средства. В том числе проводят экспериментальные хирургические методы лечения: паллидотомия и таламотомия. Однако большинство больных, перенесших оперативное вмешательство, возвращаются к изначальному уровню проявлений уже через год после операции.
В целом неблагоприятный, так как болезнь неизлечима. Сроки наступления инвалидизации зависят от клинической формы заболевания. Наиболее благоприятный прогноз — при позднем дебюте заболевания. При этом деменция выражена слабо, симптомы прогрессируют медленно.
При ранней манифестации проявления устойчивы и прогрессируют быстро. Полная потеря способности к самообслуживанию происходит через 10 лет от начала заболевания.
Не стоит пренебрегать лечением. Оно улучшает качество жизни пациента, дает возможность отсрочить момент инвалидизации.
Чем могут помочь современные врачи?
В современной медицине не существует методов лечения, которые могут предотвратить или остановить болезнь Галлервордена-Шпатца.
Терапия направлена на облегчение и снятие интенсивности симптоматики:
- При синдроме паркинсонизма назначают агонисты дофамина (Проноран, Прамипексол, Мирапекс, Пирибедил), амантадины (Симметрел, Мидантан). Однако часто наблюдается резистентность синдрома к лечению.
- Для купирования гиперкинезов используют вальпроаты (Конвулекс, Депакин, Энкорат), бензодиазепины (Клоназепам, Диазепам).
- Для снятия спастичности мышц применяются миорелаксанты (Мидокалм, Баклофен).
- Эпилептические приступы снимаются вальпроатами, Томапаксом.
- При когнитивных нарушениях применяют Глиатилин, Нейромидин.
- Для лечения психических нарушений рекомендуют прием нейролептиков (Клоназепама, Кветиапина, Рисполента), антидепрессантов (Дапоксетина, Циталопрама, Венлафаксина).
Появляются и новейшие методы лечения болезни. К ним относятся терапия путем введения пантотеновой кислоты, магнитная стимуляция головного мозга (бледного шара).
Прогноз
Болезнь Галлервордена — Шпатца характеризуется неуклонным прогрессированием симптомов. Наиболее агрессивное течение свойственно детской форме: через 6-15 лет после проявления патологии наступает полная инвалидизация.
Поздний вариант болезни имеет более благоприятный прогноз, особенно если сопровождается маловыраженной деменцией. Благодаря терапии степень симптомов можно снизить, и сохранить способность пациента к самообслуживанию. Средняя продолжительность атипичной формы заболевания – 20 и больше лет.
Литература
- Hochspringen ↑ Zhou u.a.: A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. in: Nature genetics (Nat Genet.). New York 28.2001,4 (Aug), 345—349. PMID 11479594 ISSN 1061-4036
- Hochspringen ↑ PKAN-Artikel bei genereviews von Gregory/Hayflick, revidierte Version Jan. 2008
- Hochspringen ↑ M. Shevell: Hallervorden and history. in: The New England journal of medicine (N Engl J Med.). Waltham Mass 2.2003,1 (Jan),348,3-4. PMID 12510036 ISSN 0028-4793
Эта страница в последний раз была отредактирована 26 декабря 2019 в 12:14.
Общая информация
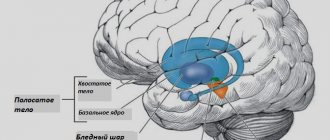
Базальные ганглии – нейронные подкорковые узлы, находящиеся в центральном белом веществе полушарий. Они состоят из полосатого тела, стриатума и чечевицеобразного ядра. В структуру последнего входят бледный шар и скорлупа. Кроме того, к базальным ганглиям относится часть черной субстанции – звена экстрапирамидной системы.
Предназначение базальных ганглиев:
- регуляция двигательных и вегетативных функций (дыхания, работы сердечно-сосудистой системы);
- участие в интегративных процессах высшей нервной деятельности.
При болезни Галлервордена — Шпатца в бледном шаре и черном веществе накапливается железо, что приводит к нарушению их работы. Как следствие, возникают моторные дисфункции и психические расстройства. Впервые заболевание было описано в 1922 году медиками Юлиусом Галлерворденом и Хуго Шпатцем. Оно встречается у 3 человек из 1000000.
Протекание заболевания
Обычное начало заболевания в детском возрасте, иногда с выраженной симптоматикой уже на первом году жизни. Редко возможна манифестация во взрослом возрасте. Сначала возникают экстрапирамидные моторные нарушения, в особенности нарушения ходьбы с тенденцией к падениям или дистонией ног, реже — психические нарушения.
В дальнейшем — нарушения движений (дистонии, хореоатетоз, тремор) с ригидностью затылочной мускулатуры, гиперрефлексией и нарушениями психики (как правило, расстройство интеллекта в виде прогрессирующей деменции). Также обнаруживаются дизартрия и дисфагия. Можно обнаружить пигментацию сетчатки или атрофию зрительного нерва[2].
Смерть наступает обычно в возрасте до 30 лет, в состоянии полного психического распада личности.
Из-за связей Юлиуса Галлервордена с программой эвтаназии в фашистской Германии было предложено называть заболевание «нейродегенерация с отложением железа в мозге 1» (англ. neurodegeneration with brain iron accumulation 1). В мире распространено сокращённое название заболевания как «NBIA1».