Causes of Leber's hereditary optic neuropathy
The disease often progresses in patients aged 12 to 25 years.
In this case, the hereditary factor plays a significant role in the occurrence of the disease. Clinically, the disease is similar to bilateral retrobulbar neuritis. Within two days, sudden loss of vision develops in both eyes, sometimes the sharpness decreases first in one and then in the other organ.
https://www.youtube.com/watch?v=ytcopyrightde
Over the next two weeks, the quality of vision continues to fall, and then stops at a certain level. Complete blindness is relatively rare.
A characteristic feature of Leber's hereditary optic neuropathy is incomplete penetrance (up to 40% in men and 15% in women) and a high incidence of damage among men (they get sick 5 times more often than women). This may be due to the effect of an X-linked modifying gene located in the Xp21 region.
The most common causes of the disease are:
- infectious inflammation of the central nervous system and optic nerves;
- congenital and acquired hydrocephalic pathologies;
- cranial oncology;
- cerebral paralysis;
- metabolic disorders;
- intoxication (lead, drugs, mercury);
- congenital and genetic pathologies of the optic nerve.
The following factors influence the development of the disease:
- stress;
- smoking and drinking alcohol;
- exposure to toxins;
- some medications and infections.
The causes of Leber neuropathy, in which the optic nerve atrophies, are genetic. The disease, caused by a specific mutation in mitochondrial genes, is transmitted by women to their offspring. Men and women inherit pathology from their mother. Why is this happening? A huge amount of DNA is found in the cell nucleus and only a small part of it is in the mitochondria.
In a significant proportion of people who are carriers of the mutated gene, the disease will not become apparent. More than 85% of women and 50% of men who are carriers of the LHON syndrome gene will not be affected by the disease. The reasons influencing the progression of the disease are not clear, but it is reliably known that the disease can be triggered by unfavorable environmental conditions, stress, infections, and the toxic effects of tobacco and alcohol.
Treatment
The disease is dangerous because the destroyed nerve fiber cannot be regenerated.
The effect of therapy can only be due to the restoration of the functioning of fibers that were functional at the time of exposure.
Treatment involves normalizing blood circulation
and
stimulation of vital processes in oppressed nerve fibers
. For this purpose, vasodilating drugs are used, drugs that improve trophic properties and also stimulate the central nervous system.
Important!
Treatment of the disease is more effective when the drug remains in the damaged area for a long time.
To obtain the maximum effect numerous injections are needed
, and this is quite painful.
Irrigation therapy
will allow fractional administration of drugs. Medicines are delivered through a catheter, which is inserted into the retrobulbar space through an opening in the skin in the lower corner of the orbit. The catheter is closed with a sterile stopper and fixed to the skin with adhesive tape.
In young children, the process is performed under inhalation mask anesthesia. For older people - under local. Medicines are administered 5-6 times a day
by piercing the catheter plug with a syringe needle after pre-treatment with ethyl alcohol.
The doctor selects a set of medications depending on the stage of the disease. The treatment period is 7-10 days.
Leber syndrome is a rare congenital disorder associated with visual impairment. The basis is a violation of cellular organelles, mitochondria. The disease occurs in 1 person in several tens of thousands of healthy people.
Leber's hereditary optic atrophy = Leber hereditary optic neuropathy (LHON) is a rare hereditary disease that causes visual impairment. The disease most often occurs between the ages of 27 and 34 and predominantly affects men.
The disease was first diagnosed by the German ophthalmologist Albrecht von Graeff in 1858, but was named after his assistant Theodor Leber, who later described the clinical course of the disease in 15 patients. Leber atrophy is the first disease associated with maternal inheritance and a specific point mutation in mitochondrial DNA (mtDNA).
Diagnosis of the disease is difficult due to the low incidence, indicating the presence of this disorder in the family. An ophthalmological examination is necessary to exclude other causes of visual impairment. It is advisable to perform a genetic study to confirm the mutation.
Symptoms of Leber's hereditary optic atrophy
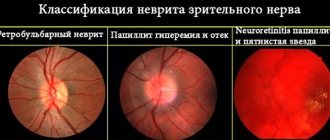
In the initial stages of the disease, the fundus of the eye remains unchanged, sometimes only some hyperemia of the optic papillae and blurring of the boundaries are noted. When diagnosing visual fields, central scotomas are observed.
Photo 1. This is what the fundus looks like in the normal state of the organ of vision (on the left) and with atrophy of the optic nerve (on the right).
Atrophy is classified into several types:
- simple (primary) and secondary (post-inflammatory or post-congestive) - the first is characterized by normal vision loss, narrowing of the lateral visual field;
- partial and complete type - complete or partial loss of vision;
- stationary or progressive - with the first type, the process of vision loss stops at some stage, and with a progressive form, a gradual decline in visual function is observed, which can lead to complete atrophy of the nerve, i.e., blindness;
- unilateral and bilateral type - damage to one or two eyes.
Reference. Most patients experience progressive deterioration of vision over months or even years, but approximately 20% of patients experience improvement in vision. There are cases of complete restoration of vision.
The list of symptoms of optic nerve atrophy is quite extensive and depends on the type of pathology. The most common symptoms of all types of pathology:
- decreased visual acuity;
- violation of accommodation;
- night blindness.
In an advanced state of the disease, the general symptoms of damage to the visual pathways are accompanied by signs indicating damage to the central nervous system. This includes cases:
- complicated dementia;
- depression;
- the appearance of bulbar symptoms;
- ataxia of the cerebellar and spinal type;
- spastic paraplegia.
https://www.youtube.com/watch?v=https:accounts.google.comServiceLogin
In such situations, differential diagnosis is carried out to exclude the risk of multiple sclerosis, tumor of the optic nerve or chiasmatic region.
Attention! The disease develops at a young age (usually from 12 to 25 years), so any signs of visual impairment should not be ignored.
Any visual impairment that rapidly progresses must be carefully studied and comprehensively diagnosed. Leber's optic neuropathy is rarely accompanied by malaise characteristic of neurological diseases, and vision declines quickly, sometimes not even within a few months, but within two to three weeks.
With such acuity, vision cannot be checked using Sivtsev’s table, because a person simply does not see the signs, so they use hardware diagnostics and the “counting fingers” method at a certain distance from the patient’s face. The peculiarity of the disease is that the optic nerve is affected due to a gene mutation. An excess amount of toxic oxygen molecules is produced, which negatively affects the condition of nerve cells and vision decreases.
Leber's optic neuropathy - symptoms and features of the disease:
- young age of patients - 18-35 years;
- the disease develops more often in men;
- rapid unilateral and then bilateral vision loss;
- visual acuity decreases over several weeks;
- violation of color perception of red and green colors;
- the pathology may be accompanied by symptoms inherent in mitochondrial diseases (convulsions, cardiac conduction disturbances).
With Leber optic nerve atrophy, most often the process of disease development begins in one eye, but there are often cases when two eyes are affected at once. Central vision is constantly decreasing. This is due to the gradual death of cells in the optic nerve, which transmits image information to the brain.
While hereditary optic neuropathy is considered an incurable disease, scientists are constantly searching for new ways to treat such a pathology. Thanks to gene therapy methods, it is possible to significantly slow down the progression of diseases and improve the quality of life of patients.
The rapid progression of Leber's disease leads to rapid loss of vision in people who have seen normally all their lives and had no vision problems. A disappointing diagnosis, as well as information that the disease has not yet been treated, seriously affects the well-being of such patients who were not prepared for disability. Many of the existing methods of treating pathology have proven to be ineffective, including surgical intervention.
Fortunately, scientists involved in rare pathologies are working hard to find a cure. Experts place particular hope in gene therapy, and many research groups have already achieved good results. Sometimes an obstacle to continuing experiments is their ethical side, since further research involves the use of the developed technique on humans. Are there any successes at least using experimental models? Yes, geneticists have already managed to find a way to solve the problem.
Thus, a group of scientists from Miami, using experimental models, proved that mutated genes can be safely replaced with healthy ones, this will prevent deterioration in the nutrition of optic nerve cells. To correct a genetic defect in the mitochondria, it is necessary to introduce normal DNA - this will correct the disorder and restore visual function.
Genetics: how, to whom and when the disease is transmitted
Mitochondrial inheritance pattern of Leber syndrome
Leber's hereditary optic atrophy is mediated by a DNA mutation in the mitochondria, which a person (mostly male) always receives from the mother, since only the egg cell transfers its mitochondria to the developing embryo (mitochondria from the father's sperm are not transferred).
Although the vast majority of patients with Leber disease have homoplasmic mutations, 10-15% of mutations are heteroplasmic. Tissue-specific segregation may be responsible for differences in interindividual phenotypes. Some studies suggest that the risk to patients is minimal if heteroplasm is less than 60%. Sons of mothers who have heteroplasma levels ≤80% are less likely to suffer from the disease.
The issue under discussion is the appearance of Leber syndrome in female carriers of mutations, which, depending on the genetic background, have significantly lower penetrance than males. Some studies suggest that the cause of differential penetrance is a modifying X-linked gene, leading to the manifestation of the disease in women only in the homozygous state. The second putative factor is X-inactivation of the “wild-type” X chromosome.
Diagnosis of Leber optic neuropathy
Symptoms of optic nerve atrophy appear not only in the early stages of the development of the disease, but also as a result of serious injuries to the areas of the brain that are responsible for visual functions.
When examining a patient, the doctor examines the presence of concomitant diseases, facts of taking pharmacology and contact with chemicals, addictions, as well as complaints that indicate possible intracranial lesions.
In the physical form of diagnosis, ophthalmologists determine the presence or absence of exophthalmos, examine the mobility of the eyeball, test the reaction of the pupil to light, and the corneal reflex. Be sure to check visual acuity, perimetry, and study color perception.
List of diagnostic measures:
- ophthalmoscopy - the degree of blurring of the nerve boundary is analyzed;
Photo 2. The ophthalmoscopy process: a special device directs a beam of light onto the eye, which helps to see the patient’s fundus.
- visual acuity test, determining the boundaries of the visual field;
- angiography of cerebral vessels supplying the nerve;
- identification of damaged nerve areas using computer perimetry;
- tomography;
- craniographic examination;
- VEP, which determines a decrease in lability and an increase in the sensitivity threshold of the optic nerve;
- for glaucoma, intraocular pressure is measured;
- plain radiography of the orbit of the eye - study of pathologies of the orbit;
- fluorescein angiography - examination of the retinal vascular network;
- radiography of the skull and sella turcica;
- magnetic resonance imaging (MRI) - assessment of optic nerve fibers;
- a blood test confirming or refuting the presence of an inflammatory process;
- ELISA and PCR diagnostics.
Diagnosis of the disease is complicated by the similarity of its symptoms with ischemic neuropathy or with certain pathologies - for example, multiple sclerosis. It is also difficult to track the history, since not all carriers of the mutated gene develop Leber neuropathy. An extensive ophthalmological examination can only establish the presence of pathology, but to determine the cause, genetic diagnosis will be needed.
https://www.youtube.com/watch?v=ytadvertisede
Features of diagnosing Leber hereditary optic neuropathy:
- general clinical examination;
- Mitochondrial DNA analysis;
- visual field examination;
- fundus examination;
- coherence tomography and electroretinography (necessary to exclude retinal pathologies).
Specialists are recommended to carry out a differential diagnosis of LHON syndrome by comparing symptoms and examination data with other diseases affecting the optic nerve. For example, with ischemic, toxic neuropathy. A complete ophthalmological examination shows that the optic disc is inflamed, and vascular telangiectasia (dilation of small capillaries) is also noticeable.
Prevention
Since Leber optic atrophy is a hereditary disease, its prevention is difficult. For preventive purposes, it is advisable to promptly treat problems that can cause the disorder.
The next point is to avoid any eye injuries. A healthy lifestyle, quitting smoking and drinking alcohol is also important.
There are several forms of hereditary optic nerve atrophy that differ from each other in clinical manifestations, the nature of functional disorders, the time of onset of the disease, and the type of inheritance. Treatment of hereditary optic nerve atrophy should be aimed at improving trophism; as a rule, it is ineffective.
Juvenile hereditary optic atrophy
- a bilateral disease with an autosomal dominant mode of inheritance. It occurs more often than other hereditary atrophies and is the most benign form. The first ophthalmoscopic signs appear at 2-3 years of age, functional disorders occur much later (at 7-20 years). Visual acuity decreases gradually, but remains fairly preserved for a long time, amounting to 0.1-0.9. Central and paracentral scotomas appear, and the blind spot increases. Concentric narrowing of the visual field is rarely observed. Color vision impairment usually precedes a decrease in visual acuity. First, sensitivity to blue decreases, then to red and green; Complete color blindness may develop. Dark adaptation does not change. The electroretinogram is usually normal. The disease may be accompanied by nystagmus and neurological disorders.
Congenital, or infantile, hereditary autosomal recessive optic atrophy is less common than the dominant form and usually appears at birth or at an early age (up to 3 years). Atrophy is bilateral, complete, stationary. Visual acuity is sharply reduced, the field of vision is concentrically narrowed. There is dyschromatopsia. Electroretinogram is normal. Nystagmus is usually observed. General and neurological disorders are rare. The disease should be differentiated from disc hypoplasia, an infantile form of taperetinal degeneration.
Sex-linked optic atrophy is rare, appears early in life, and progresses slowly. Visual acuity decreases to 0.4-0.1. The peripheral parts of the visual field are preserved, the blind spot is slightly enlarged. In the early stages of the disease (at a young age), the electroretinogram is normal, subsequently the b wave decreases and disappears. Optic nerve atrophy can be combined with moderate neurological impairment.
Complicated infantile hereditary atrophy of the Beer optic nerve is more often transmitted by a recessive type, less often by a dominant type. It begins early - in the 3-10th year of life, when vision suddenly decreases, then the process slowly progresses.
In the early stages of the disease, mild hyperemia of the disc is observed. Subsequently, partial (with damage to the temporal half of the disc) or complete atrophy of the optic nerve develops. Visual acuity may decrease to 0.05-0.2; Complete blindness, as a rule, does not occur. There is a central scotoma with normal boundaries of the peripheral visual field. Often combined with nystagmus (50%) and strabismus (75%). Characterized by the presence of neurological symptoms; The pyramidal system is predominantly affected, which makes this form similar to hereditary ataxias.
Atrophy
(neuritis)
of Leber's optic nerve
. It begins suddenly and proceeds as acute bilateral retrobulbar neuritis. The interval between damage to one eye and the other can sometimes reach 1-6 months. Men are more often affected (up to 80-90% of cases). The disease can appear at the age of 5-65 years, more often - at 13-28 years. Within a few days, less often 2-4 weeks, vision decreases to 0.1 - counting fingers on the face. Sometimes a decrease in vision is preceded by periods of blurring; only in isolated cases are photopsia observed. Nyctalopia is often observed; patients see better at dusk than during the day. In the initial stages of the disease, headache may occur. Central scotomas are revealed in the field of view, the periphery is often preserved, and the electroretinogram is not changed. Dychromatopsia in red and green colors is characteristic.
The fundus may be normal, sometimes there is slight hyperemia and slight blurring of the boundaries of the optic nerve head.
Atrophic changes appear 3-4 months after the onset of the disease, first in the temporal part of the disc. In the late stage, optic nerve atrophy develops.
Some patients experience relapses or experience slow progression of the process; some patients experience some improvement in visual function. Neurological disorders occur rarely. Sometimes there are deviations in the EEG, mild signs of damage to the membranes and the diencephalic region.
In members of the same family, the disease for the most part proceeds in the same way with regard to the time of its onset, the nature and degree of functional impairment. The type of inheritance has not been precisely established; transmission by a recessive type linked to sex is more likely.
Opticodiabetic syndrome
- bilateral primary optic nerve atrophy, accompanied by a sharp decrease in vision in combination with deafness of neurogenic origin, hydronephrosis, malformations of the urinary system, diabetes mellitus or diabetes insipidus. Develops between the ages of 2 and 24, most often before 15 years.
Leber hereditary optic neuropathy LHON, or Leber optic atrophy, is an inherited (passed from mother to offspring) mitochondrial degeneration of retinal ganglion cells (GGCs) and their axons, resulting in acute or near-acute loss of central vision; it affects predominantly young men.
However, LHON is transmitted only through the maternal line, primarily due to mutations (non-nuclear) in the mitochondrial genome, and only the egg contributes to the mitochondria in the embryo. LHON is typically associated with one of three pathogenic mitochondrial DNA (mtDNA) point mutations. These mutations act on nucleotides and reposition 11,778 G to A, 3,460 G to A, and 14,484 T to C, respectively, in the ND4, ND1, and Nd6 gene subunits in complex I chain oxidative phosphorylation in mitochondria. Men cannot pass the disease on to their offspring.
Leber optic atrophy lesions are limited primarily to retinal ganglion cells with preserved pigment epithelium and the photoreceptor layer. The disease reveals axonal degeneration, demyelination and atrophy of the visual pathway: from the optic nerve to the lateral geniculate bodies. It has been shown that during the disease there is a deterioration in glutamate transport with disruption of mitochondria, which leads to death and apoptosis of retinal ganglion cells. However, selective damage to individual retinal fibers has not yet been fully studied.
The disease is characterized by acute or subacute painless loss of vision caused by bilateral optic atrophy. As a rule, at the beginning of the disease, visual acuity in one eye decreases, then after a short period of time (an average of 6-8 weeks) changes in the second optic nerve occur. Pain when moving the eyeballs is not typical for this syndrome and is more common in acute optic neuritis.
In most patients, clinical manifestations are limited to optic nerve pathology. But in some pedigrees, optic nerve atrophy is combined with symptoms inherent in mitochondrial diseases (cardiac conduction disorders, extrapyramidal disorders, seizures, diabetes mellitus). Some neurological symptoms were noted in 45-60% of people with LHON. One of the relatively common symptoms is tremor; it occurs in 20% of patients.
The disease usually manifests itself at the age of 15-35 years (however, the age of onset of the disease can vary from 1 to 70 years). It is characterized by acute or subacute bilateral slow decrease in the acuity of central vision, and is not accompanied by pain in the eyeballs.
The eyes can be affected either simultaneously or sequentially, with an interval of several months. As a rule, the decrease in vision remains pronounced and constant, but cases have been described when, after a few years, a spontaneous improvement in vision occurs, sometimes significant. In the early stages of the disease, color vision is often affected. Sometimes neurological symptoms are detected: tremor, ataxia, dystonia, convulsions, in some cases the symptoms are similar to multiple sclerosis.
The disease is characterized by incomplete penetrance (up to 50% in men and 10% in women) and a higher frequency among men (men get sick 3-5 times more often than women). The provoking factor in the onset and development of the disease are risk factors - stress, smoking, alcohol consumption, effects of toxins, drugs and infections. It has been shown that the severity of the disease and the possibility of vision restoration correlate with the identified mutations. Thus, it is believed that the m.11778G>A mutation causes the most severe forms, m.3460G>A causes milder forms, and m.14484T>C gives the most favorable prognosis.
The diagnosis of NAZNL is established after a detailed examination, which includes examination of the fundus, visual field examination to identify a central scotoma, registration of visual evoked potentials to confirm the involvement of the optic nerve in the process, electroretinography to exclude retinal diseases, optical coherence tomography to identify characteristic structural changes in the nerve layer retinal fibers, neuroimaging to exclude other diseases and DNA diagnostics to verify the diagnosis.
Differential diagnosis should be carried out with other diseases affecting the optic nerve. Conventionally, all these diseases can be divided according to the pattern of visual disturbances. There are patterns of retrobulbar neuritis (RBN), ischemic neuropathy, infiltrative lesions, compression effects, toxic neuropathy and hereditary degeneration.
The literature describes individual observations indicating the effectiveness of therapy for NADNL with idebenone, a synthetic precursor of coenzyme Q10, as monotherapy and in combination with vitamins.
NALD is one of the main causes of slowly progressive bilateral painless optic atrophy. If such a pattern of visual disturbances develops, a detailed family history should be collected and DNA diagnostics should be performed to exclude NAZNL. Establishing a correct diagnosis will allow you to avoid unreasonable prescriptions, conduct pathogenetic treatment and medical genetic counseling.
Despite the fact that many of the diseases described in this section are considered incurable, the Center for the Treatment of Rare Diseases in Milan is constantly looking for new methods. Thanks to gene therapy, it has been possible to achieve outstanding results and completely cure some rare syndromes.
Contact a consultant on the website or leave a request - this way you can find out what methods Italian doctors offer. Perhaps this disease has already been treated in Milan.
Leber Hereditary Optic Neuropathy (NOHL or LHON), also known as Leber Optic Atrophy (LOA), is named after Dr. Theodore Leber, who described it in 1871 as a characteristic pattern of sudden loss of vision in young people with a family history of loss of vision was observed. It is the most common hereditary optic neuropathy and is caused by a mitochondrial mutation and its prevalence is very low and unknown in most geographic areas. In the north-east of England and Finland, the disease affects 1 in every 31,000 or 50,000 people respectively.
It is usually seen in young men aged 18 to 35, although it can also affect younger children and adults over 35. In women it is much less common.
It usually causes severe vision loss in both eyes. In most cases, the process begins in one eye and moves to the other eye after a few weeks or months.
Why is it produced? What is mitochondrial heredity?
Although the vast majority of our genetic material (DNA) is located in the nucleus of our cells, a small portion is found in the mitochondria (mitochondrial DNA or mtDNA).
Core genes are inherited from both biological parents. However, mitochondrial DNA is inherited only from the mother. This means that a man with a mutation in mitochondrial DNA will not pass it on to any of his biological descendants, whereas a woman with a mutation in mitochondrial DNA will pass it on to all of her biological descendants.
90-95% of LHON cases are associated with one of three specific mtDNA mutations. However, a significant proportion of people with these mutations never develop symptoms of the disease. Specifically, only 10% of women and 50% of men carrying any of these mutations have optic neuropathy. This implies that other factors, genetic (mitochondrial or nuclear) and/or environmental factors must act so that vision loss occurs. For example, exposure to tobacco or large amounts of alcohol in people carrying these mutations increases the risk of developing the disease.
Symptoms of Leber Optic Neuropathy
Typically, LHON carriers do not have vision until they lose vision significantly quickly in one eye and, after a few weeks or months, in the other eye as well. Vision continues to deteriorate over several weeks until it becomes significant, and in most cases central vision (needed for reading, driving, and recognizing faces) is severely affected in both eyes, although some people experience some vision recovery after some time. This permanent vision loss results from the death of the optic nerve cells responsible for transmitting images from the eyes to the brain.
Although vision loss is the only symptom in most patients with LHON, abnormal heart rhythms as well as neurological changes (such as postural tremors or other movement disorders) have been described in some cases.
How is it diagnosed? Is there a treatment for this disease?
This disease is difficult to diagnose for several reasons. First, it is a hereditary disease, but it does not affect all people who have the mutation. For this reason, it may appear without taking into account a family history of the disease.
To diagnose it, it usually requires a thorough neurophthalmologic evaluation and blood testing to examine mtDNA.
Fundus examination initially shows microangiopathy and swelling of the peripapillary nerve fiber layer, which progresses to optic atrophy.
The current prognosis is usually permanent, severe vision loss. Several treatments for the disease are being explored, and research into the disease is underway at the ICR and is in the screening phase.
Pathophysiology[edit | edit code]
Ocular pathology is limited to the retinal ganglion cell layer, especially the maculopapillary ganglion. Degeneration is evident from the ganglion cells of the retinal organs to the axonal pathways leading to the lateral lateral geniculate body. Experimental data show impaired glutamate transport and an increase in reactive oxygen species (ROS) causing apoptosis of retinal ganglion cells. In addition, experiments show that normally, without LHON, retinal ganglion cells produce less of the potent free radical superoxide than other normal neurons of the central nervous system.[18] Viral vector experiments increasing superoxide dismutase 2 in LHON cybrids[19] or LHON animal models or using exogenous glutathione in LHON cybrids[20] have shown that there is a risk of death of LHON-affected retinal ganglion cells from apoptosis. These experiments may partly explain the preference for death of LHON-affected retinal ganglion cells over other central nervous system neurons that also harbor LHON-affected mitochondria.
Notes
- Bandelt HJ, Kong QP, Parson W, Salas A (December 2005). "More evidence for non-maternal inheritance of mitochondrial DNA?". J. Med. Genet. 42
(12): 957-60. DOI:10.1136/jmg.2005.033589. PMID 15923271. - Leber T. Ueber hereditaere und congenital angelegte sehnervenleiden (1871) Graefes Arch Clin Exp Ophthalmol. 17:249-291
- Erickson R. P. (1972). "Leber's optic atrophy, a possible example of maternal inheritance." Am J Hum Genet 24
(3):348–349. - Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ 2nd, Nikoskelainen EK (1988). "Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy." Science 242
(4884):1427-1430. DOI:10.1126/science.3201231. PMID 3201231. - Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML (1991). “A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy.” Am J Hum Genet 48
(6):1147–1153. - Johns DR, Neufeld MJ, Park RD (1992). "An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy." Biochem Biophys Res Commun 187
(3):1551–1557. DOI:10.1016/0006-291x(92)90479-5. - Nikoskelainen EK, Marttila RJ, Huoponen K, et al. (August 1995). "Leber's "plus": neurological abnormalities in patients with Leber's hereditary optic neuropathy." J. Neurol. Neurosurg. Psychiatr. 59
(2): 160-4. DOI:10.1136/jnnp.59.2.160. PMID 7629530. - cardiac arrythmia
- Mayo Clinic: Multiple Sclerosis
- OMIM 535000
- Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF (February 2003). "The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England". Am. J.Hum. Genet. 72
(2): 333-9. DOI:10.1086/346066. PMID 12518276. - Puomila A, Hamalainen P, Kivioja S, et al. (October 2007). "Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland". Eur. J.Hum. Genet. 15
(10): 1079-89. DOI:10.1038/sj.ejhg.5201828. PMID 17406640. - Laberge AM, Jomphe M, Houde L, et al. (2005). "A "Fille du Roy" Introduced the T14484C Leber Hereditary Optic Neuropathy Mutation in French Canadians." Am. J.Hum. Genet. 77
(2): 313-7. DOI:10.1086/432491. PMID 15954041. - Hudson G, Carelli V, Horvath R, Zeviani M, Smeets HJ, Chinnery PF (2007). "X-Inactivation patterns in females harboring mtDNA mutations that cause Leber hereditary optic neuropathy." Mol. Vis. 13
: 2339-43. PMID 18199976. - Hudson G, Keers S, Yu Wai Man P, et al. (December 2005). "Identification of an X-Chromosomal Locus and Haplotype Modulating the Phenotype of a Mitochondrial DNA Disorder". Am. J.Hum. Genet. 77
(6): 1086-91. DOI:10.1086/498176. PMID 16380918. - Chinnery PF, Andrews RM, Turnbull DM, Howell NN (January 2001). "Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation?". Am. J. Med. Genet. 98
(3): 235-43. DOI:10.1002/1096-8628(20010122)98:3<235::AID-AJMG1086>3.0.CO;2-O. PMID 11169561. - Hudson G, Carelli V, Spruijt L, et al. (August 2007). "Clinical Expression of Leber Hereditary Optic Neuropathy Is Affected by the Mitochondrial DNA-Haplogroup Background." Am. J.Hum. Genet. 81
(2): 228-33. DOI:10.1086/519394. PMID 17668373. - Hoegger MJ, Lieven CJ, Levin LA (2008). "Differential production of superoxide by neuronal mitochondria". BMC Neurosci 9
: 4. DOI:10.1186/1471-2202-9-4. PMID 18182110. - ↑ 1 2
Qi X, Sun L, Hauswirth WW, Lewin AS, Guy J (February 2007).
"Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutation." Arch. Ophthalmol. 125
(2):268–72. DOI:10.1001/archopht.125.2.268. PMID 17296905. - ↑ 1 2
Ghelli A, Porcelli AM, Zanna C, Martinuzzi A, Carelli V, Rugolo M (February 2008).
"Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids." Invest. Ophthalmol. Vis. Sci. 49
(2): 671–6. DOI:10.1167/iovs.07-0880. PMID 18235013. - GeneTests LHON search (inaccessible link)
- ↑ 1 2
Klopstock, T.;
Yu-Wai-Man, P., Dimitriadis, K., Rouleau, J., Heck, S., Bailie, M., Atawan, A., Chattopadhyay, S., Schubert, M., Garip, A., Kernt , M., Petraki, D., Rummey, C., Leinonen, M., Metz, G., Griffiths, P.G., Meier, T., Chinnery, P.F. (25 July 2011). "A randomized placebo-controlled trial of idebenone in Leber's hereditary optic neuropathy." Brain 134
(9): 2677. DOI:10.1093/brain/awr170. - ↑ 1 2 Shrader WD, Amagata A., Barnes A., Enns GM, Hinman A., Jankowski O., Kheifets V., Komatsuzaki R., Lee E., Mollard P., Murase K., Sadun AA, Thoolen M ., Wesson K., Miller G.
α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. (English) // Bioorganic & medicinal chemistry letters. - 2011. - Vol. 21, no. 12. - P. 3693—3698. — DOI:10.1016/j.bmcl.2011.04.085. - PMID 21600768. [correct] - Sadun AA, Salomao SR, Berezovsky A, et al. (2006). "SUBCLINICAL CARRIERS AND CONVERSIONS IN LEBER HEREDITARY OPTIC NEUROPATHY: A PROSPECTIVE PSYCHOPHYSICAL STUDY." Trans Am Ophthalmol Soc 104
:51–61. PMID 17471325. - Carelli V, Ross-Cisneros FN, Sadun AA (January 2004). "Mitochondrial dysfunction as a cause of optic neuropathies." Prog Retin Eye Res 23
(1):53–89. DOI:10.1016/j.preteyeres.2003.10.003. PMID 14766317. - Katz, Jason.
Parkland Manual of Inpatient Medicine. - Dallas, TX: FA Davis, 2006. - P. 903. - Clinical Idebenone trial recruiting at Newcastle University UK https://lhon.ncl.ac.uk
- Mashima Y, Kigasawa K, Wakakura M, Oguchi Y (September 2000). "Do idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy?". J Neuroophthalmol 20
(3):166–70. DOI:10.1097/00041327-200020030-00006. PMID 11001192. - Archived copy (unavailable link - history
). Retrieved June 5, 2011. Archived September 4, 2011. Sadun,A et al. "EPI-743 alters the natural history of progression of Leber hereditary optic neuropathy." AOS meeting. May 2011] - Newman NJ, Biousse V, David R, et al. (September 2005). "Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite." Am. J. Ophthalmol. 140
(3):407–15. DOI:10.1016/j.ajo.2005.03.058. PMID 16083844. - Haroon MF, Fatima A, Scholer S, et al. (2007). "Minocycline, a possible neuroprotective agent in Leber's hereditary optic neuropathy (LHON): Studies of cybrid cells bearing 11778 mutation." Neurobiol Dis 28
(3):237–50. DOI:10.1016/j.nbd.2007.07.021. PMID 17822909. - Clinical Curcurmin trial recruiting at ClinicalTrials.nlm.nih.gov Archived February 13, 2009.
- Wisconsin near infrared trial Archived May 15, 2008.
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, Herbert M, Turnbull DM (May 2010). "Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease." Nature 465
(7294):82–85. DOI:10.1038/nature08958. PMID 20393463.