Причины наследственной оптической нейропатии Лебера
Недуг зачастую прогрессирует у пациентов в возрасте от 12 до 25 лет. При этом весомую роль в возникновении заболевания играет наследственный фактор. По клиническим показателям болезнь схожа с двусторонним ретробульбарным невритом.
В течение двух дней развивается внезапная потеря зрения на обоих глазах, иногда острота снижается сперва на одном, а потом и на другом органе.
https://www.youtube.com/watch?v=ytcopyrightde
В течение последующих двух недель качество зрения продолжает падать, а затем останавливается на определённом уровне. Полная слепота появляется относительно редко.
Характерная особенность наследственной оптической нейропатии Лебера — неполная пенетрантность (до 40% у мужчин и 15% у женщин) и высокая частота поражения среди мужчин (они заболевают в 5 раз чаще, чем женщины). Это, возможно, связано с воздействием Х-сцепленного модифицирующего гена, находящегося в районе Xp21.
Наиболее частыми причинами развития недуга считаются:
- инфекционные воспаления ЦНС и зрительных нервов;
- врожденные и приобретенные гидроцефалические патологии;
- онкология черепной коробки;
- церебральный паралич;
- метаболические нарушения;
- интоксикации (свинцом, лекарственными препаратами, ртутью);
- врожденные и генетические патологии зрительного нерва.
Влияние на развитие заболевания оказывают следующие факторы:
- стресс;
- курение и употребление спиртных напитков;
- воздействие токсинов;
- некоторые лекарства и инфекции.
Причины нейропатии Лебера, при которой атрофируется зрительный нерв, — генетические. Болезнь, обусловленная специфической мутацией генов митохондрий, передается женщинами своему потомству. Мужчины и женщины наследуют патологию от матери. Почему так происходит? Огромное количество ДНК находится в ядре клеток и лишь небольшая его часть — в митохондриях.
У значительной части людей, которые являются носителем мутированного гена, болезнь не станет явной. Более 85% женщин и 50% мужчин, которые являются носителем гена синдрома LHON, болезнь не затронет. Причины, влияющие на прогрессирование заболевания, не ясны, но достоверно известно, что спровоцировать болезнь может неблагоприятная экологическая обстановка, стрессы, инфекции, токсическое воздействие табака и алкоголя.
Лечение
Недуг опасен тем, что разрушенное нервное волокно не подлежит регенерации.
Эффект от терапии может быть обусловлен только восстановлением функционирования дееспособных на момент воздействия волокон.
Лечение подразумевает нормализацию кровообращения
и
стимулирование процессов жизнедеятельности в угнетенных нервных волокнах
. С этой целью применяются сосудорасширяющие лекарства, препараты, которые улучшают трофические свойства, а также стимулируют ЦНС.
Важно!
Лечение недуга более результативно при длительном нахождении лекарственного вещества в зоне повреждения. Для получения максимального эффекта
нужны многочисленные инъекции
, а это довольно болезненно.
Ирригационная терапия
позволит дробно вводить лекарственные средства. Медикаменты поступают через катетер, который устанавливается в ретробульбарное пространство через отверстие в коже в нижнем углу глазницы. Катетер закрывают стерильной пробкой и фиксируют пластырем к коже.
У маленьких детей процесс выполняют под ингаляционным масочным наркозом. У старших — под местным. Лекарственные препараты вводят 5—6 раз в день
при помощи прокалывания иглой шприца пробки катетера после предварительной обработки этиловым спиртом. Набор препаратов подбирает врач в зависимости от стадии недуга. Срок лечения
7—10 дней.
Синдром Лебера – это редкое врожденное заболевание, ассоциирующееся с нарушениями зрения. В основе лежит нарушение клеточных органелл, митохондрий. Заболевание встречается у 1 человека на несколько десятков тысяч здоровых людей.
Наследственная Лебера (англ.: Leber optic atrophy = Leber hereditary optic neuropathy, LHON) – это редкое наследственное заболевание, вызывающее нарушения зрения. Болезнь чаще всего встречается в возрасте 27-34 лет, преимущественно поражает мужчин.
Болезнь была впервые диагностирована немецким офтальмологом Альбрехтом фон Граефом в 1858 г, но название получила в честь его ассистента Теодора Лебера, позже описавшего клиническое течение заболевания у 15 пациентов. Атрофия Лебера – первое заболевание, связанное с материнской наследственностью и специфической точечной мутацией в митохондриальной ДНК (мтДНК).
Диагностика болезни затруднена из-за низкой заболеваемости, свидетельствующей о наличии этого расстройства в семье. Для исключения других причин нарушения зрения необходимо офтальмологическое обследование. Целесообразно выполнение генетического исследования для подтверждения мутации.
Симптомы наследственной атрофии зрительного нерва Лебера
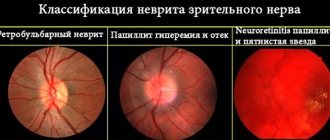
На начальных стадиях заболевания глазное дно остается без изменений, иногда отмечают лишь некоторую гиперемию сосочков зрительных нервов и размытие границ. При диагностике полей зрения наблюдаются центральные скотомы.
Фото 1. Так выглядит глазное дно при нормальном состоянии органа зрения (слева) и при атрофии зрительного нерва (справа).
Атрофию классифицируют на несколько типов:
- простой (первичный) и вторичный (поствоспалительный или послезастойный) — первый характеризуется обычной потерей зрения, сужением бокового зрительного поля;
- частичный и полный тип — полная или частичная потеря зрения;
- стационарный или прогрессирующий — при первой разновидности процесс потери зрения на какой-то стадии останавливается, а при прогрессирующей форме наблюдается постепенный спад зрительной функции, который может привести к полной атрофии нерва, т. е. к слепоте;
- односторонний и двусторонний тип — поражение одного или двух глаз.
Справка. У большинства пациентов обнаруживают прогрессивное ухудшение зрения в течение месяцев и даже лет, но примерно у 20% больных отмечают улучшение зрения. Известны случаи полного восстановления зрения.
Перечень симптомов атрофии зрительного нерва довольно обширен и зависит от вида патологии. Самые распространённые симптомы всех типов патологии:
- снижение остроты зрения;
- нарушение аккомодации;
- куриная слепота.
В запущенном состоянии болезни к общим симптомам повреждения зрительных путей присоединяются признаки, свидетельствующие о поражении ЦНС. Сюда относятся случаи:
- осложнённой деменции;
- депрессии;
- появления бульбарных симптомов;
- атаксии мозжечкового и спинального типа;
- спастической параплегии.
https://www.youtube.com/watch?v=https:accounts.google.comServiceLogin
В таких ситуациях проводят дифференциальную диагностику, чтобы исключить риск возникновения рассеянного склероза, опухоли зрительного нерва или хиазмальной области.
Внимание! Заболевание развивается в молодом возрасте (чаще от 12 до 25 лет), поэтому любые признаки нарушения зрительной функции не должны оставаться без внимания.
Любые нарушения зрения, стремительно прогрессирующие, должны быть тщательно изучены и комплексно продиагностированы. Оптическая нейропатия Лебера редко сопровождается недомоганием, характерным для неврологических заболеваний, при этом зрение падает быстро, иногда даже не в течение нескольких месяцев, а за две-три недели.
При такой остроте, зрение невозможно проверить по таблице Сивцева, потому что человек попросту не видит знаков, поэтому применяют аппаратную диагностику и метод «счета пальцев» на определенном расстоянии от лица пациента. Особенность заболевания в том, что зрительный нерв поражается из-за мутации гена. Вырабатывается избыточное количество токсичных молекул кислорода, что негативно сказывается на состоянии клеток нерва и зрение снижается.
Оптическая нейропатия Лебера — симптомы и особенности болезни:
- молодой возраст пациентов — 18-35 лет;
- болезнь чаще развивается у мужчин;
- быстрая односторонняя, а затем и двусторонняя потеря зрения;
- острота зрения снижается в течение нескольких недель;
- нарушение цветового восприятия красного и зеленого цветов;
- патология может сопровождаться симптомами, присущими митохондриальным болезням (судороги, нарушение проводимости сердца).
При атрофии зрительного нерва Лебера чаще всего процесс развития болезни начинается с одного глаза, но нередки случаи, когда поражаются сразу два глаза. Центральное зрение снижается постоянно. Это связано с постепенной смертью клеток зрительного нерва, передающего информацию об изображении в головной мозг.
Пока наследственная нейропатия оптическая считается неизлечимым заболеванием, но учеными ведется постоянный поиск новых путей для терапии подобной патологии. Благодаря методам генной терапии можно существенно замедлить прогрессирование болезней и улучшить качество жизни пациентов.
Стремительное прогрессирование болезни Лебера приводит к быстрой потере зрения людьми, которые всю свою жизнь видели нормально и не имели проблем со зрением. Неутешительный диагноз, а также информация о том, что болезнь пока не лечится, серьезно сказываются на самочувствии таких пациентов, которые не были готовы к инвалидности. Многие из существующих методик лечения патологии оказались малоэффективными, в том числе и хирургическое вмешательство.
К счастью, ученые, занимающиеся редкими патологиями, прикладывают немало усилий к тому, чтобы найти средство излечения. Особую надежду специалисты возлагают на генную терапию, и у многих исследовательских групп уже есть неплохие результаты. Иногда препятствием к продолжению экспериментов является их этическая сторона, так как дальнейшие исследования предполагают использование разработанной методики на людях. Есть ли успехи хотя бы с использованием экспериментальных моделей? Да, генетикам уже удалось найти путь к решению проблемы.
Так, группа ученых из Майами, используя экспериментальные модели, доказала, что мутировавшие гены можно безопасно заменить здоровыми, это предотвратит ухудшение питания клеток зрительного нерва. Для исправления генетического дефекта в митохондрии необходимо ввести нормальную ДНК — это позволит исправить нарушение и восстановить зрительную функцию.
Генетика: как, кому и когда передается болезнь
Митохондриальный шаблон наследования синдрома Лебера
Наследственная атрофия зрительных нервов Лебера опосредуется мутацией ДНК в митохондриях, которую человек (преимущественно, мужского пола) всегда получает от матери, поскольку только яйцеклетка передает свои митохондрии зарождающемуся эмбриону (митохондрии отцовской спермы не передаются).
Хотя подавляющее большинство пациентов с болезнью Лебера имеют гомоплазматические мутации, 10-15% мутаций – гетероплазматические. За отличия в межиндивидуальных фенотипах может быть ответственна тканеспецифичная сегрегация. Некоторые исследования показывают, что риск для пациентов минимален, если гетероплазма составляет менее 60%. Сыновья матерей, у которых уровни гетероплазмы ≤80%, с меньшей вероятностью будут страдать от болезни.
Обсуждаемый вопрос – появление синдрома Лебера у женщин-носителей мутаций, которые, в зависимости от генетического фона, имеют значительно меньшую пенетрантность, чем мужчины. Некоторые исследования предполагают, что причина дифференцированной пенетрантности – модифицирующий Х-связанный ген, приводящий к проявлению заболевания у женщин только в гомозиготном состоянии. Второй предполагаемый фактор – Х-инактивация «wild-type» Х-хромосомы.
Диагностика оптической нейропатии Лебера
Симптомы атрофии зрительного нерва появляются не только на первых порах развития недуга, но и в результате получения серьезных травм участков головного мозга, которые ответственны за зрительные функции.
При осмотре пациента врач изучает наличие сопутствующих заболеваний, факты приема фармакологии и контакта с химическими препаратами, пагубные привычки, а также жалобы, которые свидетельствуют о возможных интракраниальных поражениях.
При физикальной форме диагностики офтальмологи определяют присутствие либо отсутствие экзофтальма, исследуют подвижность глазного яблока, тестируют реакцию зрачка на свет, рефлекс роговицы. Обязательно проводят проверку остроты зрения, периметрию, изучение цветовосприятия.
Перечень диагностических мер:
- офтальмоскопия — анализируется степень размытия границы нерва;
Фото 2. Процесс офтальмоскопии: специальный прибор направляет пучок света на глаз, что помогает увидеть глазное дно пациента.
- тест на остроту зрения, определение границы зрительного поля;
- ангиография сосудов головного мозга, питающих нерв;
- выявление повреждённых участков нерва при помощи компьютерной периметрии;
- томография;
- краниографическое исследование;
- ЗВП, определяющее снижение лабильности и увеличение порога чувствительности зрительного нерва;
- при глаукоме измеряют внутриглазное давление;
- обзорная рентгенография орбиты глаза — изучение патологий глазницы;
- флуоресцентная ангиография — осмотр сосудистой сетки сетчатки;
- рентгенография черепа и турецкого седла;
- магнитно-резонансная томография (МРТ) — оценка волокон зрительного нерва;
- анализ крови, подтверждающий или опровергающий наличие воспалительного процесса;
- ИФА и ПЦР-диагностика.
Диагностика болезни осложнена схожестью ее симптомов с ишемической невропатией или с определенными патологиями — например, с рассеянным склерозом. Сложно отследить и анамнез, так как не у всех носителей мутировавшего гена развивается нейропатия Лебера. Расширенное офтальмологическое обследование может только установить наличие патологии, а вот для определения причины, понадобится генетическая диагностика.
https://www.youtube.com/watch?v=ytadvertisede
Особенности диагностики наследственной оптической нейропатии Лебера:
- общее клиническое обследование;
- анализ митохондриальной ДНК;
- исследование полей зрения;
- осмотр глазного дна;
- когерентная томография и электроретинография (необходимы, чтобы исключить патологии сетчатки).
Специалистам рекомендуется проводить дифференциальную диагностику синдрома LHON, сравнивая симптоматику и данные обследования с другими заболеваниями, поражающими глазной нерв. Например, с ишемической, токсической невропатией. Полное офтальмологическое обследование показывает, что диск зрительного нерва воспаленный, также заметны телеангиэктазии сосудов (расширение мелких капилляров).
Профилактика
Поскольку атрофия зрительного нерва Лебера – это наследственная болезнь, ее профилактика сложная. В превентивных целях целесообразно вовремя лечить проблемы, способные стать причиной расстройства.
Следующий пункт – избегание любых , глазных травм. Важен также здоровый образ жизни, отказ от курения, потребления алкоголя.
Известно несколько форм наследственных атрофии зрительного нерва, отличающихся друг от друга клиническими проявлениями, характером функциональных нарушений, временем начала заболевания, типом наследования. Лечение наследственных атрофии зрительного нерва должно быть направлено на улучшение трофики; как правило, оно малоэффективно.
Юношеская наследственная атрофия зрительного нерва
— двустороннее заболевание с аутосомно-доминантным типом наследования. Встречается чаще других наследственных атрофии и является наиболее доброкачественной формой. Первые офтальмоскопические признаки появляются в 2-3 летнем возрасте, функциональные нарушения наступают значительно позже (в 7-20 лет). Острота зрения снижается постепенно, длительное время остается достаточно сохранной, составляя 0,1-0,9. Появляются центральные и парацентральные скотомы, увеличивается слепое пятно. Концентрическое сужение поля зрения наблюдается редко. Нарушения цветового зрения, как правило, предшествуют снижению остроты зрения. Сначала снижается чувствительность к синему цвету, затем — к красному и зеленому; может развиться полная цветослепота. Темновая адаптация не изменяется. Электроретинограмма, как правило, в норме. Заболевание может сопровождаться нистагмом и неврологическими нарушениями.
Врожденная, или инфантильная, наследственная аутосомно-рецессивная атрофия зрительного нерва встречается реже, чем доминантная форма, проявляется, как правило, при рождении или в раннем возрасте (до 3 лет). Атрофия двусторонняя, полная, стационарная. Острота зрения резко снижена, поле зрения концентрически сужено. Имеется дисхроматопсия. Электроретинограмма в норме. Обычно наблюдается нистагм. Общие и неврологические расстройства встречаются редко. Заболевание следует дифференцировать от гипоплазии диска, инфантильной формы тапеторетинальной дегенерации.
Атрофия зрительного нерва, связанная с полом, встречается редко, проявляется в раннем возрасте и медленно прогрессирует. Острота зрения снижается до 0,4-0,1. Периферические отделы поля зрения сохранены, слепое пятно несколько увеличено. В ранних стадиях заболевания (в молодом возрасте) электроретинограмма в норме, впоследствии снижается и исчезает волна b. Атрофия зрительного нерва может сочетаться с умеренными неврологическими нарушениями.
Осложненная инфантильная наследственная атрофия зрительного нерва Бера чаще передается по рецессивному типу, реже — по доминантному. Начинается рано — на 3-10-м году жизни, когда внезапно снижается зрение, затем процесс медленно прогрессирует.
В ранних стадиях заболевания наблюдается легкая гиперемия диска. Впоследствии развивается частичная (с поражением височной половины диска) или полная атрофия зрительного нерва. Острота зрения может снижаться до 0,05-0,2; полная слепота, как правило, не наступает. Имеется центральная скотома при нормальных границах периферического поля зрения. Часто сочетается с нистагмом (50%) и косоглазием (75%). Характерно наличие неврологических симптомов; поражается преимущественно пирамидная система, что сближает эту форму с наследственными атаксиями.
Атрофия
(неврит)
зрительного нерва Лебера
. Начинается внезапно и протекает по типу острого двустороннего ретробульбарно-го неврита. Интервал между поражением одного и другого глаза иногда может достигать 1-6 мес. Чаще болеют мужчины (до 80-90% случаев). Заболевание может появляться в возрасте 5-65 лет, чаще — в 13-28 лет. В течение нескольких дней, реже 2 -4 нед, зрение снижается до 0,1 — счет пальцев у лица. Иногда снижению зрения предшествуют периоды затуманивания, лишь в единичных случаях наблюдаются фотопсии. Нередко отмечается никталопия, больные лучше видят в сумерки, чем днем. В начальном периоде заболевания может отмечаться головная боль. В поле зрения выявляются центральные скотомы, периферия чаще сохранена, электроретинограмма не изменена. Характерна дисхроматопсия на красный и зеленый цвета.
Глазное дно может быть нормальным, иногда отмечается легкая гиперемия и небольшая нечеткость границ диска зрительного нерва.
Атрофические изменения появляются через 3-4 мес после начала заболевания, сначала в височной части диска. В поздней стадии развивается атрофия зрительного нерва.
У некоторых больных возникают рецидивы или наблюдается медленное прогрессирование процесса, у ряда больных отмечается некоторое улучшение зрительных функций. Неврологические расстройства возникают редко. Иногда отмечаются отклонения на ЭЭГ, нерезко выраженные признаки поражения оболочек и диэнцефальной области.
У членов одной семьи заболевание большей частью протекает однотипно в отношении времени его начала, характера и степени функциональных нарушений. Тип наследования точно не установлен, более вероятна передача по рецессивному типу, сцепленному с полом.
Оптикоотодиабетичеекий синдром
— двусторонняя первичная атрофия зрительного нерва, сопровождающаяся резким снижением зрения в сочетании с глухотой неврогенного генеза, гидронефрозом, пороками развития мочевой системы, сахарным или несахарным диабетом. Развивается в возрасте от 2 до 24, чаще до 15 лет.
Наследственная оптическая нейропатия LHON Лебера, или атрофия зрительного нерва Лебера, является наследственной (передаётся от матери к потомству) митохондриальной дегенерацией ганглионарных клеток (РСК) сетчатки и их аксонов, что приводит к острой или почти острой потере центрального зрения; это влияет преимущественно на молодых мужчин.
Тем не менее, LHON передается только по материнской линии, прежде всего, из-за мутаций (не ядерных) в митохондриальном геноме, и только яйцеклетка способствует митохондрии в зародыше. LHON, как правило, связана с одной из трех патогенных митохондриальных ДНК (мтДНК) точечных мутаций. Эти мутации действуют на нуклеотиды и репозиционируют 11778 G в А, 3460 G в А и 14484 T в C , соответственно, в ND4, ND1 и Nd6 субъединицах генов в комплексе I окислительного фосфорилирования цепочек в митохондриях. Мужчины не могут передать болезнь своему потомству.
Нарушения при атрофии зрительных нервов Лебера ограничиваются преимущественно ганглиозными клетками сетчатки с сохраненным пигментным эпителием и слоя фоторецепторов. При заболевании обнаруживают аксональную дегенерацию, демиелинизацию и атрофию зрительного пути: от зрительного нерва до латеральных коленчатых тел. Показано, что при болезни происходит ухудшение транспорта глутамата с нарушением работы митохондрий, что приводит к гибели и апоптозу ганглиозных клеток сетчатки. Однако, избирательное повреждение отдельных волокон сетчатки пока до конца не изучено.
Заболевание характеризуется острой или подострой безболезненной потерей зрения, вызванное билатеральной атрофией зрительного нерва. Как правило, вначале заболевания снижается острота зрения на один глаз, затем через небольшой промежуток времени (в среднем 6-8 недель) присоединяются изменения второго зрительного нерва. Боли при движении глазных яблок не характерны для данного синдрома и чаще встречаются при остром неврите зрительного нерва.
У большинства больных клинические проявления ограничиваются патологией зрительного нерва. Но в некоторых родословных атрофия зрительных нервов сочетается с симптомами, присущими митохондриальным болезням (нарушение сердечной проводимости, экстрапирамидные нарушения, судороги, сахарный диабет). Те или иные неврологические симптомы отмечены у 45-60% лиц с LHON . Одним из относительно часто встречающихся симптомов является тремор, он встречается у 20% больных.
Заболевание манифестирует, как правило, в возрасте 15-35 лет (однако возраст начала заболевания может варьировать от 1 до 70 лет). Характеризуется острым или подострым двусторонним медленным снижением остроты центрального зрения, при этом не сопровождается болью в глазных яблоках.
Глаза могут поражаться как одновременно, так и последовательно, с интервалом в несколько месяцев. Как правило, снижение зрения остается выраженным и постоянным, но описаны случаи, когда спустя несколько лет происходит спонтанное улучшение зрения, иногда значительное. На ранних стадиях заболевания часто отмечается поражение цветового зрения. Иногда выявляются неврологические симптомы: тремор, атаксии, дистония, судороги, в некоторых случаях симптоматика схожа с рассеянным склерозом.
Заболевание характеризуется неполной пенетрантностью (до 50% у мужчин и 10% у женщин) и большей частотой среди мужчин (мужчины болеют в 3-5 раз чаще женщин), Провоцирующим фактором начала и развития заболевания являются факторы риска — стрессы, курение, употребление алкоголя, действие токсинов, лекарств и инфекций. Показано, что степень тяжести заболевания и возможность восстановления зрения коррелируют с выявленными мутациями. Так, считается, что мутация m.11778G>A вызывает наиболее тяжелые формы, m.3460G>A — более легкие, а m.14484T>C дает наиболее благоприятный прогноз.
Диагноз НАЗНЛ устанавливается после подробного обследования, в которое входит исследование глазного дна, исследование полей зрения для выявления центральной скотомы, регистрация зрительных вызванных потенциалов для подтверждения вовлечения в процесс зрительного нерва, электроретинография для исключения заболеваний сетчатки, оптическая когерентная томография для выявления характерных структурных изменений слоя нервных волокон сетчатки, нейровизуализация для исключения других заболеваний и ДНКдиагностика для верификации диагноза.
Дифференциальную диагностику следует проводить с другими заболеваниями, поражающими зрительный нерв. Условно все эти заболевания могут быть разделены в соответствии с паттерном зрительных нарушений. Выделяют паттерн ретробульбарного неврита (РБН), ишемической невропатии, инфильтративного поражения, компрессионного воздействия, токсической невропатии и наследственной дегенерации.
В литературе описаны отдельные наблюдения, указывающие на эффективность терапии НАЗНЛ идебеноном, синтетическим предшественником коэнзима Q10, в качестве монотерапии и в сочетании с витаминами.
НАЗНЛ является одной из основных причин медленно прогрессирующей двусторонней безболевой атрофии зрительных нервов. При развитии подобного паттерна зрительных нарушений должен быть собран подробный семейный анамнез и проведена ДНК-диагностика для исключения НАЗНЛ. Постановка верного диагноза позволит избежать необоснованных назначений, провести патогенетическое лечение и медико-генетическое консультирование.
Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку — так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
Наследственная Оптическая Нейропатия Лебера (NOHL или LHON), также известная как Атрофия Зрительного Нерва Лебера (LOA), названа в честь доктора Теодора Лебера, который описал её в 1871 году, как характерную картину внезапной потери зрения у молодых людей, в с семейном анамнезе которых наблюдалась потеря зрения. Это самая распространенная наследственная оптическая невропатия, её причиной является митохондриальная мутация и её распространенность очень низкая, их в большинстве географических районов она неизвестна. На северо-востоке Англии и Финляндии заболевание затрагивает 1 человека из каждых 31 000 или 50 000 соответственно.
Это обычно наблюдается у молодых мужчин в возрасте от 18 до 35 лет, хотя это может также повлиять на детей младшего возраста и взрослых старше 35 лет. У женщин оно встречается гораздо реже.
Как правило, оно вызывает серьезную потерю зрения в обоих глазах. В большинстве случаев, процесс начинает в одном глазу и через несколько недель или месяцев переходит на второй глаз.
Почему оно производится? Что такое наследственность митохондрий?
Хотя подавляющее большинство нашего генетического материала (ДНК) расположено в ядре клеток, его небольшая часть находится в митохондриях (митохондриальная ДНК или мтДНК).
Гены ядра наследуются от обоих биологических родителей. Однако, ДНК митохондрии наследуются только от матери. Это означает, что мужчина с мутацией в митохондриальной ДНК не передаст его ни одному из своих биологических потомков, тогда как женщина с мутацией в митохондриальной ДНК передаст её всем своим биологическим потомкам.
90-95% случаев LHON связаны с одной из трех специфических мутаций мтДНК. Однако значительная часть людей с этими мутациями никогда не развивают симптомы заболевания. В частности, только 10% женщин и 50% мужчин, несущих любую из этих мутаций, имеют оптическую нейропатию . Это подразумевает, что другие факторы, генетические (митохондриальные или ядерные) и / или факторы окружающей среды должны действовать так, чтобы произошла потеря зрения. Так, например, воздействие табака или большого количества алкоголя у людей, несущих эти мутации, увеличивает риск развития заболевания.
Симптомы Оптической Нейропатии Лебера
Как правило, носители LHON не имеют до тех пор, пока они не теряют значительно быстро зрение в одном глазу и через несколько недель или месяцев также в другом глазу. Зрение продолжает ухудшаться в течение нескольких недель, пока оно не станет значительным, и в большинстве случаев центральное видение (необходимое для чтения, вождения и распознавания лиц) серьезно страдает в обоих глазах, хотя некоторые люди испытывают определенное восстановление зрения через некоторое время. Эта постоянная потеря зрения является следствием смерти клеток зрительного нерва, ответственных за передачу изображений из глаз в головной мозг.
Хотя потеря зрения является единственным симптомом у большинства пациентов с LHON, в некоторых случаях из них были описаны нарушения сердечного ритма, а также неврологические изменения (такие как постуральный тремор или другие нарушения движения).
Как оно диагностируется? Существует лечение этого заболевания?
Это заболевание сложно диагностировать по нескольким причинам. Во-первых, это наследственное заболевание, но оно не проявляется у всех людей, имеющих мутации. По этой причине он может появиться без учета семейного анамнеза болезни.
Чтобы диагностировать его, обычно необходимо провести тщательную нейрофтальмологическую оценку и анализ крови для исследования мтДНК.
В анализе глазного дна сначала наблюдается микроангиопатия и отек слоя перипапиллярного нервного волокна, который прогрессирует до оптической атрофии.
В настоящее время прогноз обычно представляет собой постоянную серьезную потерю зрения. Изучается несколько способов лечения этого заболевания, и в ICR проводится исследование болезни, которая находится на этапе отбора.
Патофизиология[править | править код]
Глазная патология ограничивается слоем ганглиозных клеток сетчатки особенно макулопапилярного узла. Вырождение очевидно от ганглиозных клеток органов сетчатки до аксональных путей, ведущих к боковому латеральному коленчатому телу . Экспериментальные данные показывает нарушение транспорта глутамата и увеличение активных форм кислорода (АФК), вызывающие апоптоз ганглиозных клеток сетчатки. Кроме того, опыты показывают, что обычно, без LHON, ганглиозные клетки сетчатки производят меньше сильнодействующего свободнорадикального супероксида, чем другие обычные нейроны центральной нервной системы.[18] В экспериментах вирусного вектора, увеличивающих супероксиддисмутаза 2 в LHON цибридах[19] или LHON моделей животных или использования экзогенного глутатиона в LHON цибридах[20] было показано, что существует риск гибели пострадавших от LHON ганглиозных клеток сетчатки от апоптоза. Эти эксперименты могут частично объяснить предпочтительность смерти пострадавших от LHON ганглиозных клеток сетчатки другим нейронам центральной нервной системы, которые также несут пострадавшие от LHON митохондрии.
Примечания
- Bandelt HJ, Kong QP, Parson W, Salas A (December 2005). «More evidence for non-maternal inheritance of mitochondrial DNA?». J. Med. Genet.42
(12): 957-60. DOI:10.1136/jmg.2005.033589. PMID 15923271. - Leber T. Ueber hereditaere und congenital angelegte sehnervenleiden (1871) Graefes Arch Clin Exp Ophthalmol. 17:249-291
- Erickson RP (1972). «Leber’s optic atrophy, a possible example of maternal inheritance». Am J Hum Genet24
(3): 348-349. - Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ 2nd, Nikoskelainen EK (1988). «Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy». Science242
(4884): 1427-1430. DOI:10.1126/science.3201231. PMID 3201231. - Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML (1991). «A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy». Am J Hum Genet48
(6): 1147-1153. - Johns DR, Neufeld MJ, Park RD (1992). «An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy». Biochem Biophys Res Commun187
(3): 1551-1557. DOI:10.1016/0006-291x(92)90479-5. - Nikoskelainen EK, Marttila RJ, Huoponen K, et al. (August 1995). «Leber’s «plus»: neurological abnormalities in patients with Leber’s hereditary optic neuropathy». J. Neurol. Neurosurg. Psychiatr.59
(2): 160-4. DOI:10.1136/jnnp.59.2.160. PMID 7629530. - cardiac arrythmia
- Mayo Clinic: Multiple Sclerosis
- OMIM 535000
- Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF (February 2003). «The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England». Am. J. Hum. Genet.72
(2): 333-9. DOI:10.1086/346066. PMID 12518276. - Puomila A, Hamalainen P, Kivioja S, et al. (October 2007). «Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland». Eur. J. Hum. Genet.15
(10): 1079-89. DOI:10.1038/sj.ejhg.5201828. PMID 17406640. - Laberge AM, Jomphe M, Houde L, et al. (2005). «A «Fille du Roy» Introduced the T14484C Leber Hereditary Optic Neuropathy Mutation in French Canadians». Am. J. Hum. Genet.77
(2): 313-7. DOI:10.1086/432491. PMID 15954041. - Hudson G, Carelli V, Horvath R, Zeviani M, Smeets HJ, Chinnery PF (2007). «X-Inactivation patterns in females harboring mtDNA mutations that cause Leber hereditary optic neuropathy». Mol. Vis.13
: 2339-43. PMID 18199976. - Hudson G, Keers S, Yu Wai Man P, et al. (December 2005). «Identification of an X-Chromosomal Locus and Haplotype Modulating the Phenotype of a Mitochondrial DNA Disorder». Am. J. Hum. Genet.77
(6): 1086-91. DOI:10.1086/498176. PMID 16380918. - Chinnery PF, Andrews RM, Turnbull DM, Howell NN (January 2001). «Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation?». Am. J. Med. Genet.98
(3): 235-43. DOI:10.1002/1096-8628(20010122)98:3<235::AID-AJMG1086>3.0.CO;2-O. PMID 11169561. - Hudson G, Carelli V, Spruijt L, et al. (August 2007). «Clinical Expression of Leber Hereditary Optic Neuropathy Is Affected by the Mitochondrial DNA-Haplogroup Background». Am. J. Hum. Genet.81
(2): 228-33. DOI:10.1086/519394. PMID 17668373. - Hoegger MJ, Lieven CJ, Levin LA (2008). «Differential production of superoxide by neuronal mitochondria». BMC Neurosci9
: 4. DOI:10.1186/1471-2202-9-4. PMID 18182110. - ↑ 12
Qi X, Sun L, Hauswirth WW, Lewin AS, Guy J (February 2007). «Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutation».
Arch. Ophthalmol.125
(2): 268–72. DOI:10.1001/archopht.125.2.268. PMID 17296905. - ↑ 12
Ghelli A, Porcelli AM, Zanna C, Martinuzzi A, Carelli V, Rugolo M (February 2008). «Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids».
Invest. Ophthalmol. Vis. Sci.49
(2): 671–6. DOI:10.1167/iovs.07-0880. PMID 18235013. - GeneTests LHON search (недоступная ссылка)
- ↑ 12
Klopstock, T.; Yu-Wai-Man, P., Dimitriadis, K., Rouleau, J., Heck, S., Bailie, M., Atawan, A., Chattopadhyay, S., Schubert, M., Garip, A., Kernt, M., Petraki, D., Rummey, C., Leinonen, M., Metz, G., Griffiths, P. G., Meier, T., Chinnery, P. F. (25 July 2011). «A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy».
Brain134
(9): 2677. DOI:10.1093/brain/awr170. - ↑ 12Shrader W. D., Amagata A., Barnes A., Enns G. M., Hinman A., Jankowski O., Kheifets V., Komatsuzaki R., Lee E., Mollard P., Murase K., Sadun A. A., Thoolen M., Wesson K., Miller G.
α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. (англ.) // Bioorganic & medicinal chemistry letters. — 2011. — Vol. 21, no. 12. — P. 3693—3698. — DOI:10.1016/j.bmcl.2011.04.085. — PMID 21600768. [исправить] - Sadun AA, Salomao SR, Berezovsky A, et al. (2006). «SUBCLINICAL CARRIERS AND CONVERSIONS IN LEBER HEREDITARY OPTIC NEUROPATHY: A PROSPECTIVE PSYCHOPHYSICAL STUDY». Trans Am Ophthalmol Soc104
: 51–61. PMID 17471325. - Carelli V, Ross-Cisneros FN, Sadun AA (January 2004). «Mitochondrial dysfunction as a cause of optic neuropathies». Prog Retin Eye Res23
(1): 53–89. DOI:10.1016/j.preteyeres.2003.10.003. PMID 14766317. - Katz, Jason.
Parkland Manual of Inpatient Medicine. — Dallas, TX : FA Davis, 2006. — P. 903. - Clinical Idebenone trial recruiting at Newcastle University UK https://lhon.ncl.ac.uk
- Mashima Y, Kigasawa K, Wakakura M, Oguchi Y (September 2000). «Do idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy?». J Neuroophthalmol20
(3): 166–70. DOI:10.1097/00041327-200020030-00006. PMID 11001192. - Архивированная копия (недоступная ссылка — история
). Проверено 5 июня 2011. Архивировано 4 сентября 2011 года. Sadun,A et al. «EPI-743 alters the natural history of progression of Leber hereditary optic neuropathy». AOS meeting. May 2011] - Newman NJ, Biousse V, David R, et al. (September 2005). «Prophylaxis for second eye involvement in leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite». Am. J. Ophthalmol.140
(3): 407–15. DOI:10.1016/j.ajo.2005.03.058. PMID 16083844. - Haroon MF, Fatima A, Scholer S, et al. (2007). «Minocycline, a possible neuroprotective agent in Leber’s hereditary optic neuropathy (LHON): Studies of cybrid cells bearing 11778 mutation». Neurobiol Dis28
(3): 237–50. DOI:10.1016/j.nbd.2007.07.021. PMID 17822909. - Clinical Curcurmin trial recruiting at ClinicalTrials.nlm.nih.gov Архивировано 13 февраля 2009 года.
- Wisconsin near infrared trial Архивировано 15 мая 2008 года.
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, Herbert M, Turnbull DM (May 2010). «Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease». Nature465
(7294): 82–85. DOI:10.1038/nature08958. PMID 20393463.