Aicardi syndrome is a rare cerebroretinal genetic disorder. With this disease, the brain is partially or completely deprived of one of its main components – the corpus callosum. This is a consequence of a defect in the X chromosome. Changes occur in the retina leading to infantile spasms.
The corpus callosum is the central section of the nervous system, represented by nerve endings connecting the hemispheres of the brain.
Aicardi syndrome is not a separate form of epilepsy, but is an independent disease, which was described in 1965 by French neurologist Jean Ecardi.
Aicardi-Gutierrez syndrome is a threat to boys with the XXY chromosome set or Klinefelter syndrome.
According to statistics, to date there have been about 500 cases of this disease. It is most widespread in Japan.
However, nationality and race do not play a key role. It all depends on the level of medicine in the region. Lack of knowledge leads to undetected cases. Of the 2–4% of children who exhibit infantile spasms, Ecardi disease is detected.
Inheritance
Aicardi-Gutierrez syndrome is an inherited (autosomal recessive) condition, meaning that the disease is passed on genetically. An autosomal recessive condition occurs when both copies of a gene or gene cluster are defective.
If the fetus inherits defective copies from both parents, the disease occurs. Alternatively, the fetus may inherit one defective gene from one parent and the remaining normal copy becomes damaged due to environmental causes such as viral infection or exposure to DNA-damaging agents.
A recessive condition indicates that the disease occurs only when both copies of the gene are defective.
In the dominant form of inheritance, only one defective copy can cause a problem.
Autosomal disease indicates that the condition is passed from one generation to the next through one of the 23 chromosomes (autosomes) rather than the sex chromosomes (X or Y).
Aicardi syndrome is observed in infants within 4 months after birth and leads to physical and mental retardation. Affects the brain, skin, and immune system of the body. The immune response is similar to the body's response to a viral infection, known as "mimicking congenital infection".
At birth, typical characteristics are neurological abnormalities, increased levels of liver proteins in the blood, enlarged liver and spleen (hepatosplenomegaly), and decreased blood platelet levels (thrombocytopenia).
There are two types of Aicardi syndrome based on the stage of occurrence. The early stage of the disease is observed in newborns and infants. Late-stage disease occurs in older children (1 to 2 years of age) with less brain damage.
Clinic
What do children with Aicardi syndrome look like? Symptoms are initially quite nonspecific because babies are born at term, vaginally, and appear healthy. Up to three months, newborns develop without clinical manifestations, then epileptic seizures appear. They are presented in the form of retropulsive symmetrical serial clonic-tonic convulsions of the limbs. In the vast majority of cases, this is also accompanied by infantile spasms of the flexor muscles. In rare cases, the onset of the disease occurs in the first month of life. Epileptic seizures cannot be controlled with medication.
In the neurological status of such children, there is a decrease in the size of the skull, a decrease in muscle tone, but at the same time unilateral disinhibition of reflexes or, conversely, paresis and paralysis. In addition, half of the patients have skeletal developmental anomalies: vertebral agenesis or absence of ribs. This leads to pronounced scoliotic changes in posture.
Causes
As mentioned earlier, Aicardi Syndrome is a genetic condition that is inherited in an autosomal recessive manner. In very rare cases, the syndrome is predominantly inherited. The mutation may occur suddenly or sporadically.
The exact gene that causes it is not entirely understood. Genes that show mutations in this disorder: SAMHD1, RNaseH2A, RNaseH2B, RNaseH2C, TREX1.
The SAMHD1 gene generates a protein that is involved in the immune response. Defects lead to immune failure.
TREX1 is located on chromosome 3, called AGS1. TREX1 encodes a DNase III protein that cuts single-stranded DNA. RNaseH2A, RNaseH2B, RNaseH2C encode enzymes that form a protein complex called RNAse2H that destroy RNA. TREX1, RNaseH2A, RNaseH2B, RNaseH2C are genes that code for enzymes called nucleases.
Find out more Amnestic syndromes: types, diagnosis, treatment methods
They are involved in the destruction of DNA and RNA. When there are defects in nucleases, there are excess DNA and RNA molecules that trick the body into thinking there are viral particles present. This causes an immune reaction (the body's defense) against itself.
The consequences of the reaction are lesions observed on the skin and brain defects (encephalopathy). ADAR6 mutations are also a cause of the disorder.
RNaseH2C (AGS3) mutations on chromosome 11 are observed in 40% of cases of Aicardi-Goutières syndrome. RNaseH2B (AGS2) on chromosome 13 is observed in 14%, RNaseH2A on chromosome 19 (AGS4) – 4% of cases. TREX1 mutations – 25% of cases.
TREX1, RNaseH2A, RNaseH2B contribute to the development of early Aicardi syndrome. RNaseH2B promotes late.
There are other genetic disorders that occur due to mutations. Genetic disorders associated with mutations are listed below.
Genetic disorder caused by ADAR mutation:
- Dyschromatosis symbolrica hereditaria 1 (DSH).
Genetic disorders caused by TREX1:
- Familial biliary lupus;
- Critical encephalitis;
- Systemic lupus erythematosus (SLE);
- Autosomal dominant retinal vaginalopathy with cerebral leukodystrophy (RVCL).
Symptoms
In early-onset Aicardi-Goutiers syndrome, symptoms begin approximately 4 months after birth. The pregnancy is normal and the baby does not show any symptoms at birth. The initial signs at this stage are presented below:
- Difficulty feeding;
- Severe irritability;
- Frequent fevers for no reason;
- Erroneous sleep patterns;
- Seizures or epilepsy.
There are defects in brain development, which leads to slower head growth, deterioration of fine motor skills, and generally slower neurological development.
These symptoms are described below:
- Fear reaction even to mild stimuli;
- Only partial head control;
- Abnormalities of eye movements and infant reflexes (eg, sucking);
- Language communication also deteriorates.
These symptoms persist for several months and then stabilize or disappear. There is a cessation of disease progression.
In the late onset phase of Aicardi-Goutier Syndrome, these symptoms appear only 1 or 2 years after normal physical and brain development.
In 20% of cases, signs appear in newborns.
The uterus may show the typical features of an enlarged liver and spleen, elevated levels of liver enzymes in the blood, abnormal brain formation, decreased blood platelets (thrombocytopenia), and anemia.
Other affected organs are skin, liver, eyes, hormone and immune system deficiencies.
- Skin lesions, inflammation, scarring of tissue (eg, ears, toes, fingers);
- Glaucoma;
- Insulin-dependent diabetes mellitus;
- Antidiuretic hormone deficiency (hormonal urination);
- anemia;
- Antibodies against the immune system (autoantibodies);
- Small head (microcephaly);
- Involuntary muscle contractions affecting posture and movement.
Prevalence
Since its official recognition, Aicardi syndrome has been encountered by neurologists about five thousand times. Most patients lived in Japan and other Asian countries. According to Swiss researchers, the incidence of the disease is 15 cases per hundred thousand children. Unfortunately, statistics most likely provide underestimates, since most patients remain unexamined.
The disease develops in girls. And doctors suggest that it is precisely this that causes most cases of delayed psychophysiological development and infantile spasms.
Diagnostics
Aicardi-Goutieres syndrome can be diagnosed based on clinical symptoms using computed tomography (CT), magnetic resonance imaging (MRI), and gene analysis.
There are certain characteristic features that can be used to specifically diagnose people with Aicardi-Goutier syndrome. They are as follows:
- Increased concentration of neopterin in cerebrospinal fluid (CSF);
- More ganglion calcification in the brain;
- Increased concentration of interferon-alpha (IFN-a);
- Cerebral atrophy;
- Interferon signature;
- Changes in white matter;
- An increase in the number of white blood cells (leukocytosis).
CT scans help visualize brain calcification. Brain atrophy, changes in white matter, leukocytosis, and other changes are detected using MRI.
Find out more Stages of adaptation syndrome
Quantitative PCR is used to detect the interferon signature. This is a very reliable test because it can detect the expression of interferon-stimulated genes in the blood.
The genes are visible even after the symptoms of Aicardi-Goutieres syndrome have subsided.
Neopterin concentrations in CSF are a reliable source for confirming the diagnosis.
Individual testing for the 6 main genes that cause Aicardi-Goutier syndrome is useful to confirm the diagnosis.
Multigene panel testing is more effective and reliable for confirming the diagnosis. A multigene panel analyzes all 6 genes simultaneously.
Prenatal testing includes blood sampling (PUBS), preimplantation genetic diagnosis, genetic testing. Blood is tested on the fetus in the third trimester.
For preimplantation genetic diagnosis, families who are suspected of having causative genes for Aicardi syndrome use a gene test for the embryo.
International Neurological Journal 5 (75) 2020
The article was published on p. 182
Jean Aicardi, clinician, researcher and educator, died on August 3, 2020 at the age of 88.
Professor Aicardi devoted his entire life to child neurology and clinical epileptology. He received his scientific degree in 1955 (Faculty of Medicine of the University of Paris), was a research fellow at Harvard Medical School, headed the pediatric neurological department at the Necker University Children's Hospital in Paris, and directed research at the French National Institute of Health and Medical Research (INSERM). ) (1986–1991), was Emeritus Professor of Child Neurology at the Institute of Children's Health (London, UK) (1992–1998).
Jean Aicardi is a pioneer of pediatric neurology who has made significant contributions to the description of several neurological diseases, including Aicardi syndrome (1969); Aicardi-Gutierrez syndrome (1984); Rett syndrome (together with Bengt Hagberg); alternating hemiplegia of childhood, etc. He published 259 articles in major international peer-reviewed journals, more than 120 book chapters, and was the author or co-author of three internationally recognized books: Aicardi's Epilepsy in Children (LWW, 3rd edition with A. Arzimanoglou and R. Guerrini, 2004); Diseases of the Nervous System in Childhood (Mac Keith Press; 3rd edition, 2009; 4th edition, in press); Movement Disorders in children (together with Emilio Fernandez-Alvarez; Mac Keith Press, 2001).
He has received a number of academic honors and awards, including the Hauer Prize of the American Society of Child Neurology in 1986, the Epilepsy Research Award of the American Epilepsy Society in 1995, the Ramón y Cajal Award, the ILAE-IBE Envoy Award epilepsy and the ILAE-IBE Lifetime Achievement Award. As an educator, he believed in what he called the younger generation, and in 1999 he easily accepted the invitation to become the founder and editor-in-chief of the journal on epilepsy and its electroclinical symptoms - Epileptic Disorders, which today is the educational journal of ILAE.
Jean Aicardi treated everyone with respect; he was always ready to help, to help with thoughtful and modest advice to his colleagues and students, whom he always deeply respected, and to sick children for whom he cared so much. A tireless physician and teacher, Monsieur Aicardi will be remembered not only as one of the founders of pediatric neurology, but also as a mentor to more than 100 pediatric neurologists around the world. Many of us will remember his clinical rounds. He was the one who taught us "that a major part of the assessment, and one that is often neglected, is to observe the spontaneous activity of the child... This is the best way to assess the functions of the central nervous system as well as behavior." He firmly believed, and was right, that in this era of ubiquitous technology, careful observation of clinical signs and symptoms and their correct interpretation, based on in-depth knowledge, remain more important than ever. Thank you, monsieur!
Alexis Arzimanoglou,
professor, student of J. Aicardi,
editor-in-chief of Epileptic magazine
Disorders
From the editors of the International Neurological Journal
I was personally lucky enough to listen to Professor J. Aicardi’s lectures and attend his super-excellent clinical conferences. The main feature of Professor J. Aicardi was respect for children and their parents and the deepest respect for colleagues. Of course, his contribution to global child neurology is enormous. And this is confirmed by the two-volume book “Children’s Neurology” published in English in 2012 (650 and 680 pp.), which was translated into Russian in 2014 and will undoubtedly become a reference book for the modern thinking child neurologist.
It will be extremely interesting and useful to read the article by J. Aicardi “My Circuitous Path to Becoming a French Child Neurologist and Epileptologist” (in the Journal of Child Neurolog. 2013 Mar; 28(3): 409-415) about his path in child neurology and epileptology.
Pediatric neurologists of Ukraine deeply mourn the death of an excellent professional and person, Professor J. Aicardi.
Editor-in-Chief of the International
neurological journal", pediatric neurologist,
Professor S.K. Yevtushenko
Treatment
There are several treatment options as this is a relatively new syndrome. Research is still unraveling its details. The symptoms are treatable, but there is no cure for the disease. Based on current knowledge, the following treatments have been developed:
Reverse transcriptase inhibitor (RTI) treatment
Patients with Aicardi-Goutieres syndrome may benefit from RTI treatment because TREX1 and SAMHDI are nucleases that digest DNA. In the absence of these genes, DNA accumulation occurs, which is slowed down by reverse transcriptase inhibitors. These RTIs are used to treat patients with HIV and their safety limits are clearly known.
Antibodies against interferon alpha
Anti-interferon alpha antibodies are used to block the activity of type I interferon alpha receptor or various forms of interferon alpha in the CSF.
Anti-autoantibody drugs
Aicardi-Gouthières syndrome results in an immune response when the production of antibodies increases against the body's own cells. Medicines targeting enlarged B, T cells are a form of treatment.
Myophenolate mofetil is used against reactive T cells.
Such drugs have side effects, but their use is monitored based on the degree of progression of the disease.
Other forms of treatment
- Folinic acid treatment increases CSF folate levels.
- For an active immune response caused by Aicardi-Goutier syndrome, corticosteroid therapy is recommended.
Health Tips
Seizures can be controlled with standard procedures and medications.
Adequate attention should be paid to the method of nutrition, the type of food consumed. Adequate caloric intake must be maintained for proper development.
Appropriate treatment of chest abnormalities and physical therapy is another way to cope with the complications that arise due to lung infections in Aicardi-Goutier syndrome.
Genetic counseling is recommended in high-risk families to identify carriers of gene mutations. Infants should be regularly monitored for risk of glaucoma, diabetes, and abnormal spinal development.
Which doctor should I contact?
When a child shows typical symptoms of seizures, irritability, and erratic sleep patterns, a pediatrician should be consulted. Based on symptoms and diagnosis, the child will be referred to a neurologist.
Various terms
Cree encephalitis; AGS; familial infantile encephalopathy with chronic lymphocytosis of cerebrospinal fluid and intracranial calcification; pseudotoxicoplasmosis syndrome; encephalopathy with basal ganglia classification; pseudo-TORCH syndrome.
Lifespan
About 25% of people with Aicardi-Goutieres syndrome die before age 17.
How does the syndrome manifest itself?
Children 2 to 5 months old who have Aicardi syndrome are outwardly indistinguishable from healthy babies. Later, convulsions or infantile spasms begin to appear. They represent a type of epileptic syndrome.
The child reduces vigorous activity and falls into a stupor. The gaze is fixed on one point. The arms begin to rise up and bend.
The body arches and the legs begin to straighten sharply. This takes seconds and can be continued at any moment. During seizures, the baby becomes irritable and cries constantly. Later, such seizures often lead to epilepsy.
In parallel, other symptoms of Aicardi syndrome are present:
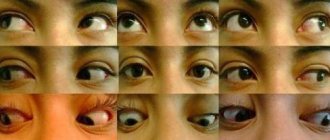
- the retina is affected, yellowish spots appear in the eyes;
The photo shows an eyeball with Aicardi syndrome - the child's eyes are abnormally small;
- congenital coloboma - a notch, gap or slit in the iris of the eye;
- developmental delay;
- problems feeding the baby;
- diarrhea;
- entry of food and gastric juice into the esophagus - gastroesophageal reflux (in adults - heartburn). It often goes away on its own and is not a serious condition. However, you should know that this is one of the signs of the disease;
- muscle lethargy.
Article on the topic: Causes of prostate adenoma - exogenous, endogenous and provoking factors
Based on observations of patients, additional signs can be identified:
- lateral location of the eyebrows, their rarity;
- protruding incisors and upturned nose;
- the angle of the nasal septum is reduced.
Sometimes growths and lumps on the skin are observed: nevi, skin diverticula and tumors, the cause of which is the pathology of blood vessels - hemangiomas. A hand anomaly is rare.
There are patients with plagiocephaly (a flattened area on the head), facial asymmetry, as well as cleft palate or upper lip. The size of the head and hands is noticeably smaller than that of a healthy person. The nose is flat and the ears are overly large. Too little space separates the nose and lips.
More patients developed severe epileptic seizures that did not go away until the end of their lives. The presence of hemivertebrae and the absence of ribs is known, leading to a noticeable curvature of the spine.
Increased risk of developing tumors:
- choroid plexus papillomas;
- lipomas;
- angisarcoma;
- hepatoblastoma;
- hemangiomas;
- angiosarcomas;
- hepatoblastoma;
- interstitial polyposis;
- embryonal calcioma;
- intestinal polyposis.
Anomalies in the eyes can lead to pigmentary renitis and microphthalmia. Cataracts and optic nerve atrophy are possible. This can lead to visual impairment and even complete blindness.
The endocrine system is often disrupted - late onset of puberty or obvious delay.
Physical development begins to slow down. At the age of 7-10 years, the patient looks like a five-year-old. Weight also increases with a delay.
According to scientists, the brain of a healthy child has more folds than that of a child with Aicardi syndrome. It happens that cysts that are filled with fluid form in the affected brain.
Most patients have a pronounced mental retardation, however, sometimes they are simply poorly able to learn.
The ability for articulate speech is often very poorly developed. Independent movement is rarely observed. Some are completely dependent on the help of other people.