Синдром Айкарди является редким цереброретинальным генетическим расстройством. При данном заболевании мозг, частично или полностью, лишен одной из основных составляющих – мозолистого тела. Это является следствием дефекта Х-хромосомы. В сетчатке происходят изменения, ведущие к инфантильным спазмам.
Мозолистое тело – центральный отдел нервной системы, представленный нервными окончаниями, соединяющими полушария головного мозга.
Сидром Айкарди не является отдельной формой эпилепсии, а является самостоятельным заболеванием, которое в 1965 году было описано невропатологом из Франции Жаном Экарди.
Синдром Айкарди-Гутьереса является угрозой для мальчиков с хромосомным набором ХХY или с синдромом Клайнфельтера.
По данным статистики, на сегодняшний момент наблюдалось около 500 случаев этого заболевания. Наибольшее распространение оно получило в Японии.
Однако, национальности и расы не играют ключевой роли. Все зависит от уровня медицины в регионе. Недостаток знаний ведет к не выявленным случаям. Из 2 – 4% детей, у которых проявляются инфантильные спазмы, обнаруживается болезнь Экарди.
Наследование
Синдром Айкарди-Гутьереса является наследственным (аутосомно-рецессивным) состоянием, что означает, что болезнь передается генетически. Автосомальное рецессивное состояние возникает, когда обе копии гена или кластера генов являются дефектными.
Если плод наследует дефектные копии от обоих родителей, то возникает заболевание. В качестве альтернативы, плод может наследовать один дефектный ген от одного родителя, а оставшаяся нормальная копия повреждается из-за экологических причин, таких как вирусная инфекция или воздействие повреждающих ДНК агентов.
Рецессивное состояние указывает на то, что заболевание возникает только в случае дефектов обеих копий гена.
При доминирующей форме наследования только одна дефектная копия может вызвать проблему.
Автосомальное заболевание указывает, что состояние передается от одного поколения к другому через одну из 23 хромосом (аутосомы), а не половых хромосом (X или Y).
Синдром Айкарди наблюдается у младенцев в течение 4 месяцев после рождения и приводит к физической, умственной отсталости. Влияет на мозг, кожу, иммунную систему организма. Иммунный ответ подобен реакции организма на вирусную инфекцию, известен как «имитация врожденной инфекции».
При рождении типичными характеристиками являются неврологические аномалии, повышенный уровень белков печени в крови, увеличение печени, селезенки (гепатоспленомегалия), снижение уровня тромбоцитов крови (тромбоцитопения).
Существует два типа синдрома Айкарди, основанных на стадии возникновения. Ранняя стадия заболевания наблюдается у новорожденных, младенцев. Поздняя стадия заболевания наблюдается у детей старшего возраста (от 1 до 2 лет) с меньшим повреждением головного мозга.
Клиника
Как же выглядят дети, имеющие синдром Айкарди? Симптомы вначале достаточно неспецифичны, потому что дети рождаются в срок, через естественные родовые пути и выглядят здоровыми. До трех месяцев новорожденные развиваются без клинических проявлений, затем появляются эпилептические приступы. Они представлены в виде ретропульсивных симметричных серийных клонико-тонических судорог конечностей. В подавляющем большинстве случаев к этому еще присоединяются инфантильные спазмы мышц-сгибателей. В редких случаях дебют заболевания встречается в первый месяц жизни. Эпилептические припадки не купируются медикаментозно.
В неврологическом статусе у таких детей наблюдается уменьшение размеров черепа, снижение тонуса мышц, но при этом одностороннее расторможение рефлексов или, наоборот, парезы и параличи. Кроме того, у половины пациентов отмечаются аномалии развития скелета: агенезия позвонков или отсутствие ребер. Это приводит к выраженным сколиотическим изменениям осанки.
Причины
Как упоминалось ранее, Синдром Айкарди является генетическим состоянием, которое наследуется аутосомно-рецессивным способом. В очень редких случаях синдром является преимущественно унаследованным. Мутация может возникать внезапно или спорадически.
Точный ген, который его вызывает, не совсем понятен. Гены, которые показывают мутации при этом расстройстве: SAMHD1, RNaseH2A, RNaseH2B, RNaseH2C, TREX1.
Ген SAMHD1 генерирует белок, который участвует при иммунном ответе. Дефекты приводят к иммунному отказу.
TREX1 расположен на хромосоме 3, называется AGS1. TREX1 кодирует белок DNase III, который режет одноцепочечную ДНК. RNaseH2A, RNaseH2B, RNaseH2C, кодируют ферменты, которые образуют белковый комплекс под названием RNAse2H, разрушают РНК. TREX1, RNaseH2A, RNaseH2B, RNaseH2C представляют собой гены, которые кодируют ферменты, называемые нуклеазами.
Узнать больше Амнестические синдромы: виды, диагностика, методы лечения
Они участвуют в разрушении ДНК и РНК. Когда есть дефекты в нуклеазах, существуют избыточные молекулы ДНК и РНК, заставляющие организм думать, что в нем присутствуют вирусные частицы. Это вызывает иммунную реакцию (защиту организма) против себя.
Последствиями реакции являются поражения, наблюдаемые на коже, дефекты головного мозга (энцефалопатия). Мутации ADAR6 также являются причиной расстройства.
Мутации RNaseH2C (AGS3) на хромосоме 11 наблюдаются в 40% случаев синдрома Айкарди-Гутьера. RNaseH2B (AGS2) на хромосоме 13 наблюдаются в 14%, RNaseH2A на хромосоме 19 (AGS4) – 4% случаев. Мутации TREX1 – 25% случаев.
TREX1, RNaseH2A, RNaseH2B способствуют развитию раннего синдрома Айкарди. RNaseH2B способствует позднему.
Существуют другие генетические нарушения, которые возникают из-за мутаций. Генетические расстройства, связанные с мутациями приведены ниже.
Генетическое расстройство, вызванное мутацией ADAR:
- Dyschromatosis symbolrica hereditaria 1 (DSH).
Генетические расстройства, вызванные TREX1:
- Семейная желчная волчанка;
- Критический энцефалит;
- Системная красная волчанка (СКВ);
- Аутосомно-доминантная вагиналопатия сетчатки с церебральной лейкодистрофией (RVCL).
Симптомы
При раннем начале синдрома Айкарди-Гутьерса симптомы появляются примерно через 4 месяца после рождения. Беременность нормальная, ребенок не проявляет никаких симптомов при рождении. Начальные признаки на этом этапе представлены ниже:
- Трудность кормления;
- Сильная раздражительность;
- Частые лихорадки без причины;
- Ошибочные режимы сна;
- Приступы или эпилепсия.
Существуют дефекты развития мозга, что приводит к замедлению роста головы, ухудшению тонких моторных навыков, вообще замедлению неврологического развития.
Эти симптомы описаны ниже:
- Реакция испуга даже на мягкие раздражители;
- Только частичное управление головкой;
- Аномалии движения глаз и детских рефлексов (например, сосание);
- Языковое общение также ухудшается.
Эти симптомы сохраняются в течение нескольких месяцев, затем стабилизируются или исчезают. Существует прекращение прогрессирования заболевания.
В фазе позднего начала Синдрома Айкарди-Гутье, эти симптомы появляются только через 1 или 2 года после нормального физического развития и развития мозга.
20% случаев признаки появляются у новорожденных.
В матке могут быть обнаружены типичные особенности увеличенной печени и селезенки, повышенные уровни ферментов печени в крови, аномальное образование головного мозга, снижение тромбоцитов крови (тромбоцитопения), анемия.
Другие пострадавшие органы – кожа, печень, глаза, дефициты гормонов и иммунной системы.
- Поражения кожи, воспаление, рубцевание ткани (например, уши, пальцы ног, пальцы);
- Глаукома;
- Инсулинзависимый сахарный диабет;
- Дефицит антидиуретического гормона (гормональное мочеиспускание);
- малокровие;
- Антитела против иммунной системы (аутоантитела);
- Малая голова (микроцефалия);
- Непроизвольные мышечные сокращения, влияющие на осанку, движение.
Распространенность
С момента своего официального признания Айкарди синдром встретился неврологам около полутысячи раз. Большинство больных проживали в Японии и других странах Азии. По данным швейцарских исследователей, встречаемость болезни — 15 случаев на сто тысяч детей. К сожалению, статистика, скорее всего, дает заниженные цифры, так как большинство пациентов остается необследованными.
Заболевание развивается у девочек. И врачи предполагают, что именно оно является причиной большинства случаев задержки психофизиологического развития и инфантильных спазмов.
Диагностика
Синдром Айкарди-Гутьера можно диагностировать на основании клинических симптомов с использованием компьютерной томографии (КТ), магнитно-резонансной томографии (МРТ), анализа генов.
Существуют определенные характерные особенности, которые могут быть использованы для конкретной диагностики людей с синдромом Айкарди-Гутье. Они заключаются в следующем:
- Увеличение концентрации неоптерина в спинномозговой жидкости (CSF);
- Больше кальцификации ганглиев в головном мозге;
- Увеличение концентрации интерферона-альфа (IFN-a);
- Церебральная атрофия;
- Интерферонная подпись;
- Изменения в белом веществе;
- Увеличение количества белых кровяных телец (лейкоцитоз).
КТ-сканирование помогает визуализировать кальцификацию головного мозга. Атрофия мозга, изменения в белом веществе, лейкоцитоз, другие изменения обнаруживаются с помощью МРТ.
Узнать больше Стадии адаптационного синдрома
Количественная ПЦР используется для обнаружения интерфероновой сигнатуры. Это очень надежный тест, поскольку может обнаруживать экспрессию интерфероно-стимулированных генов крови.
Гены видны даже после того, как симптомы синдрома Айкарди-Гутьера сократились.
Неоптериновые концентрации в CSF являются надежным источником для подтверждения диагноза.
Индивидуальное тестирование для 6 основных генов, которые вызывают синдром Айкарди-Гутье, полезно для подтверждения диагноза.
Тестирование мультигенных панелей является более эффективным и надежным для подтверждения диагноза. Мультигенная панель анализирует все 6 генов одновременно.
Пренатальное тестирование включает взятие пробы крови (PUBS), предимплантационную генетическую диагностику, генетическое тестирование. Кровь тестируется у плода в третьем триместре.
При предимплантационной генетической диагностике семьи, у которых есть подозрение о наличии причинных генов синдрома Айкарди, используют генный тест для эмбриона.
Международный неврологический журнал 5 (75) 2020
Статья опубликована на с. 182
Жан Айкарди, врач-клиницист, исследователь и педагог, скончался 3 августа 2020 года в возрасте 88 лет.
Профессор Айкарди посвятил всю свою жизнь детской неврологии и клинической эпилептологии. Он получил научную степень в 1955 году (медицинский факультет Парижского университета), был научным сотрудником в Гарвардской медицинской школе, возглавлял педиатрическое неврологическое отделение при университетском госпитале для детей Неккер в Париже, руководил научно-исследовательскими работами во Французском национальном институте здравоохранения и медицинских исследований (INSERM) (1986–1991), был почетным профессором детской неврологии в Институте здоровья детей (Лондон, Великобритания) (1992–1998).
Жан Айкарди — пионер детской неврологии, внесший значительный вклад в описание некоторых неврологических заболеваний, включая синдром Айкарди (1969); синдром Айкарди — Гутьеррес (1984); синдром Ретта (вместе с Бенгтом Хагбергом); альтернирующую гемиплегию детского возраста и др. Он опубликовал 259 статей в крупных международных рецензируемых журналах, более 120 глав из книг, был автором или соавтором трех книг, получивших международное признание: Aicardi’s Epilepsy in Children (LWW, 3rd edition with A. Arzimanoglou and R. Guerrini, 2004); Diseases of the Nervous System in Childhood (Mac Keith Press; 3rd edition, 2009; 4th edition, in press); Movement Disorders in children (together with Emilio Fernandez-Alvarez; Mac Keith Press, 2001).
Он был удостоен ряда академических званий и наград, включая Хауэровскую премию Американского общества детской неврологии в 1986 году, награду за исследования в области эпилепсии, вручаемую Американским обществом эпилепсии, в 1995 году, премию Рамон-и-Кахаля, премию посланника ILAE-IBE по вопросам эпилепсии и премию ILAE-IBE за пожизненные достижения. Как педагог он верил в тех, кого называл молодым поколением, а в 1999 году с легкостью принял приглашение стать основателем и главным редактором журнала по проблемам эпилепсии, ее электроклинической симптоматики — Epileptic Disorders, который сегодня является образовательным журналом ILAE.
Жан Айкарди относился ко всем с уважением; он готов был всегда прийти на помощь, помочь вдумчивым и скромным советом своим коллегам и студентам, которых он всегда глубоко уважал, и больным детям, о которых он так много заботился. Неутомимого врача и учителя, месье Айкарди будут помнить не только как одного из основателей детской неврологии, но и как наставника более чем 100 детских неврологов во всем мире. Его клинические обходы будут помнить многие из нас. Он был тем, кто научил нас, «что основная часть обследования и одна из тех, которыми часто пренебрегают, заключается в наблюдении за спонтанной активностью ребенка… Это лучший способ оценить функции ЦНС, а также поведение». Он твердо верил, и был прав, что в нашу эпоху повсеместного распространения технологий тщательное наблюдение за клиническими признаками и симптомами и их правильная интерпретация, основанные на глубоких знаниях, остаются важными как никогда. Спасибо, месье!
Alexis Arzimanoglou,
профессор, ученик Ж. Айкарди,
главный редактор журнала Epileptic
Disorders
От редакции «Международного неврологического журнала»
Мне лично посчастливилось слушать лекции профессора Ж. Айкарди и присутствовать на его супервеликолепных клинических конференциях. Главной чертой профессора Ж. Айкарди было уважение к детям и их родителям и глубочайшее уважение к коллегам. Безусловно, его вклад в мировую детскую неврологию огромен. И тому подтверждение — вышедший в 2012 году двухтомник на английском языке (650 и 680 с.) «Детская неврология», в 2014 году переведенный на русский язык, без сомнения, станет настольной книгой для современного думающего детского невролога.
Крайне интересно и полезно будет ознакомиться со статьей Ж. Айкарди «My Circuitous Path to Becoming a French Child Neurologist and Epileptologist» (в журнале Journal of Child Neurolog. 2013 Mar; 28(3): 409-415) о его пути в детской неврологии и эпилептологии.
Детские неврологи Украины глубоко скорбят о кончине великолепного профессионала и человека, профессора Ж. Айкарди.
Главный редактор «Международного
неврологического журнала», детский невролог,
профессор С.К. Евтушенко
Лечение
Существует несколько вариантов лечения, так как это относительно новый синдром. Исследования все еще распутывают его детали. Симптомы лечатся, однако нет лекарств от заболевания. Основываясь на текущих знаниях, были разработаны следующие методы лечения:
Обработка ингибитора обратной транскриптазы (RTI)
Пациенты с синдромом Айкарди-Гутьере могут воспользоваться обработкой RTI, поскольку TREX1 и SAMHDI являются нуклеазами, которые переваривают ДНК. В отсутствие этих генов происходит накопление ДНК, которая замедляется ингибиторами обратной транскриптазы. Эти ИРТ используются для лечения пациентов с ВИЧ, их пределы безопасности четко известны.
Антитела против интерферона альфа
Анти-интерферон-альфа-антитела используются для блокирования активности рецептора интерферона альфа-типа I или различных форм интерферона-альфа в CSF.
Препараты против аутоантител
Синдром Айкарди-Гутьера приводит к иммунному ответу, когда продуцирование антител увеличивается против собственных клеток организма. Лекарства, направленные против увеличенных В, Т-клеток, являются формой лечения.
Миофенолатный мофетил используется против реакционноспособных Т-клеток.
Такие лекарства имеют побочные эффекты, однако их использование контролируется на основе степени прогрессирования заболевания.
Другие формы лечения
- Лечение фолиновой кислотой увеличивает уровень фолата в CSF.
- При активном иммунном ответе, вызванном синдромом Айкарди-Гутье, рекомендуется терапия кортикостероидами.
Советы по здоровью
Приступы могут контролироваться стандартными процедурами и лекарствами.
Адекватное внимание следует уделять методу питания, типу потребляемой пищи. Необходимо поддерживать адекватное потребление калорий для надлежащего развития.
Подходящее лечение патологий грудной клетки и физиотерапия – еще один способ справиться с осложнениями, которые возникают из-за легочных инфекций при синдроме Айкарди-Гутье.
Генетическая консультация рекомендуется в семьях с высоким риском, чтобы выяснить наличие носителей мутаций генов. Младенцы должны регулярно контролироваться для риска появления глаукомы, сахарного диабета, неправильного развития позвоночника.
К какому врачу обратиться
Когда ребенок показывает типичные симптомы припадков, раздражительность, неустойчивые сон, следует проконсультироваться с педиатром. Основываясь на симптомах и диагнозе, ребенок будет передан неврологу.
Различные термины
Кри энцефалит; AGS; семейная инфантильная энцефалопатия с хроническим лимфоцитозом цереброспинальной жидкости и внутричерепной кальцификацией; синдром псевдотоксикоплазмоза; энцефалопатия с классификацией базальных ганглиев; синдром псевдо-TORCH.
Продолжительность жизни
Около 25% лиц с синдромом Айкарди-Гутьера умирают до 17 лет.
Как проявляет себя синдром?
Дети 2 – 5 месяцев, у которых обнаруживается синдром Айкарди, внешне не отличимы от здоровых малышей. Позже начинают проявляться судороги или инфантильные спазмы. Они представляют вид эпилептического синдрома.
Ребенок снижает активную деятельность и впадает в ступор. Взгляд прикован к одной точке. Руки начинают подниматься вверх и сгибаться.
Тело выгибается и ножки начинают резко выпрямляться. На это уходят секунды и продолжения можно ожидать в любое мгновение. Во время припадков, малыш становится раздражителен и постоянно плачет. Позже, такие судорги часто ведут к эпилепсии.
Параллельно присутствуют и другие симптомы синдрома Айкарди:
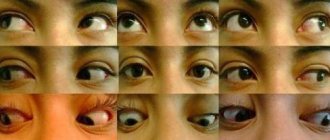
- поражается сетчатка, в глазах появляются желтоватые пятна;
На фото глазное яблоко при синдроме Айкарди - глаза ребенка ненормально-малого размера;
- врожденная колобома – выемка, щель или прорезь в радужной оболочке глаза;
- задержка развития;
- проблемы при кормлении ребенка;
- диарея;
- попадание пищи и желудочного сока в пищевод – гастроэзофагеальный рефлюкс (у взрослых-изжога). Часто проходит самостоятельно и не является серьезным заболеванием. Однако следует знать, что это один из признаков заболевания;
- мышечная апатичность.
Статья в тему: Причины аденомы простаты — экзогенные, эндогенные и провоцирующие факторы
По наблюдениям за больными можно выделить и дополнительные признаки:
- латеральное расположение бровей, их редкость;
- выступающие резцы и вздернутый нос;
- уменьшен угол носовой перегородки.
Иногда наблюдаются наросты и уплотнения на коже: невусы, дивертикулы кожи и опухоли, причиной которых является патология кровеносных сосудов – гемангиомы. Редко встречается аномалия рук.
Встречаются заболевшие с плагиоцефалией (сплюснутая область на голове), с ассиметрией лица, а также с расщелинами неба или верхней губы. Размеры головы и рук заметно меньше, чем у здорового человека. Нос плоский, а уши чрезмерно большие. Нос и губы разделяет слишком малое пространство.
У большего количества больных происходило развитие тяжелых эпилептических припадков, которые не проходили до конца жизни. Известно наличие полу-позвонков и отсутствия ребер, приводящее к заметному искривлению позвоночника.
Увеличен риск развития опухолей:
- папиллом сосудистого сплетения;
- липом;
- ангисарком;
- гепатобластом;
- гемангиом;
- ангиосарком;
- гепатобластом;
- интерстициального полипоза;
- эмбриональной кальциомы;
- полипоза кишечника.
Аномалии в глазах могут привести к пигментному рениту, микрофтальми. Возможна катаракта и атрофия зрительного нерва. Это может привести к нарушению зрения и даже полной слепоте.
Нередко нарушается эндокринная система – позднее наступление половой зрелости или явная задержка.
Физическое развитие начинает замедляться. В возрасте 7-10 лет больной выглядит как пятилетний. Вес увеличивается также с задержкой.
Согласно исследованиям ученых, в мозге здорового ребенка больше складок, чем у болеющего синдромом Айкарди. Случается, в пораженном мозге образуются кисты, которые заполнены жидкостью.
Большинство заболевших имеют явно выраженную задержку в умственном развитии, однако, иногда они просто плохо способны к обучению.
Способность к членораздельной речи часто развита очень слабо. Самостоятельное передвижение наблюдается редко. Некоторые полностью зависимы от помощи других людей.