Causes of Hallervorden-Spatz disease, risk factors
HSP is a genetic disease that is caused by an inherited defect in the pantothenate kinase 2 (PANK2) gene.
The PANK2 protein controls the formation of coenzyme A in the body. This compound helps the body convert fats, some amino acids and carbohydrates into energy. In some cases, Hallervorden-Spatz disease may not be associated with mutations in the PANK2 gene. Doctors believe that the disease is caused by a defect in several other genes. However, experts have not yet been able to identify these genes.
https://www.youtube.com/watch?v=ytpressru
Risk factors
The main risk factor is the presence of relatives in the family with this disorder. GHS usually first appears in childhood, before age 10. This form of the disease is considered early. The late form is characterized by the absence of symptoms in childhood, but their manifestation after the age of 20.
Hallervorden-Spatz disease is a genetic pathology that is both familial and sporadic. The disease is inherited in an autosomal recessive manner. The disease occurs against the background of aberrations in the pantothenate kinase gene. In total, more than 50 mutations were identified. Against the background of a genetic defect, there is a decrease in the production of pantothenate kinase, which causes the accumulation of cysteine in the basal structures, which forms stable chemical compounds with iron ions, which have a negative effect on proteins and trigger the process of peroxidation, which causes the death of neurons. In place of necrotic neurons, proliferation of glial tissue is observed.
Such pathological processes most often form in the area of the globus pallidus and substantia nigra, where extracellular iron deposits with brown pigmentation are morphologically found. In addition, there are spheroid periaxonal formations located in the white cerebral matter, cerebral cortex, spinal cord and peripheral nerve trunks.
This syndrome is associated with a mutation in the PANK2 gene, which is inherited. The transmission mechanism is autosomal recessive. This means that the mutant gene is inherited not related to sex and will only appear if there are two copies. So, there are two scenarios when a child is born with a disease:
- One of the parents is sick, and the other is a carrier.
- Both parents are carriers of the mutant gene, but do not have any clinical signs.
The PANK2 gene is responsible for the production of the enzyme pantothenate kinase 2, which is involved in the synthesis of vitamin B5 and coenzyme A. As a result of impaired enzyme synthesis in Hallervorden-Spatz disease, cysteine accumulates in the basal ganglia, and cysteine binds to iron ions. In this way, iron accumulates in the brain matter.
The reasons for the mutation are unknown. The determining factor is the transmission of the mutant gene from parents to the child. The presence of one copy of the gene (carriage) does not manifest itself clinically in any way, so it is almost impossible to predict the birth of children with the pathology.
Sad, but not hopeless
With Hallervorden-Spatz disease, symptoms constantly progress. The childhood form of the disease is the most severe. Complete disability of an individual occurs 10-15 years from the moment of the first appearance of clinical manifestations.
The most favorable development of the disease is predicted in the adult form of the disease. Especially in cases of mild dementia.
Therapy allows the patient to maintain the relative quality of life and his ability to self-care. Life expectancy with the atypical form of Hallervorden-Spatz disease can be more than 20 years.
Hereditary pallidal degeneration of Hallerwarden-Spatz (Hallerwarden-Spatz disease) is a rare hereditary degenerative disease of the nervous system, first described in 1922. To date, there are about 50 detailed descriptions of familial cases of Hallerwarden-Spatz disease (HSD) in the literature, and a number of observations have also been published sporadic cases of the disease.
Hallerwarden-Spatz disease is inherited in an autosomal recessive manner. The gene responsible for the occurrence of the disease is localized on the short arm of chromosome 20 at the 20p12.3-p13 locus. The primary molecular defect has not been established.
Pathologically, Hallervorden-Spatz disease is characterized by two main features: 1) deposition of iron-containing pigment in the area of the globus pallidus, reticular part of the substantia nigra and red nuclei, which gives them a characteristic yellowish-brown color; 2) the presence in the basal ganglia, cerebral cortex, and peripheral nerves of special spheroid neuroaxonal formations, which are local expansions of axons with proliferation of membrane and tubular structures.
Symptoms of Hallervorden-Spatz disease, diagnosis
The classic variant of Hallervorden-Spatz disease is considered to be an early childhood form with clinical manifestation between the ages of 4 and 10 years. In most cases, the disease debuts with the development of torsion dystonia, which affects the leg muscles. The main complaint is difficulty walking. Later, the process generalizes and spreads to the muscles of the pharynx, face, and trunk.
Along with generalized variants, multifocal or segmental types of dystonia may be noted. Most often, patients experience blepharospasm, facial paraspasm and spasmodic torticollis. A third of patients show signs of parkinsonism caused by muscle rigidity and hypokinesia. Sometimes the development of epileptic seizures is noted.
Hallervorden-Spatz disease is accompanied by cognitive disorders caused by a decrease in attentiveness and memory with the gradual development of oligophrenia, as well as mental disorders with a predominance of aggressiveness and antisocial behavior. Patients experience dysarthria and impaired visual acuity.
The teenage version of Hallervorden-Spatz disease manifests itself between the ages of 10 and 18 years and is due to a slower progression. The manifestation of the disease begins with the occurrence of focal torsion dystonia, most often in the muscles of the limbs or orthomandibular region. The disease is characterized by the development of mental, intellectual and behavioral disorders.
Hallervorden-Spatz disease causes a wide range of symptoms that vary depending on the severity of the disease and how far it has progressed.
A common symptom is muscle contractions that distort facial expressions and body position. Such reductions may be subject to:
- face;
- torso;
- limbs.
Other symptoms of BHS are:
- ataxia;
- convulsions;
- twisted, spasmodic, stiff limbs;
- lack of balance;
- disorientation in space;
- rave;
- inconsistency of thoughts and actions;
- stupor;
- dementia;
- slowness of gestures;
- urinary incontinence;
- salivation;
- difficulty swallowing;
- grimaces, disruption of normal facial expression;
- muscle stiffness;
- inability to form an articulate sentence;
- pain due to muscle spasms;
- incorrect body position.
The most effective methods for diagnosing Hallervorden-Spatz disease are:
- MRI;
- physical examination;
- clinical picture of the disease;
- genetic testing (possibly not in every clinic due to complexity);
- family medical history.
Since HDS is inherited, the first manifestations may already occur in childhood. However, it happens that symptoms first appear after 18 years of age.
On this basis, there are 3 variants of the course of the disease:
- Early. Manifestation before the age of 10 years. In this form, the disease progresses rapidly. The symptoms are pronounced. Impaired muscle tone makes it difficult to walk, speak, and swallow. Children quickly lose the ability to move independently.
- Teenage. A less malignant form that begins between 10 and 18 years of age. Signs of the disease are often local. Progresses more slowly.
- Late. Debut after 18 years of age is the most favorable course. Most often it is expressed by speech disorders. Over time, the clinic progresses: parkinsonism develops, mental and intellectual disorders worsen.
All forms of the disease have common symptoms. There are no specific symptoms.
https://www.youtube.com/watch?v=ytaboutru
The leading syndrome is parkinsonism. This is a whole group of neurological disorders. Parkinsonism is not the same as Parkinson's disease.
- Dystonia. One of the leading symptoms. Muscular dystonia is manifested by violent involuntary movements in the limbs. From an outside observer, the patient appears to be convulsing. Walking is significantly impaired. Dystonia can be focal:
- In the limbs. Manifests itself in dystonic postures. Characteristic for the onset of the early form of the disease and for other forms as they progress. With significant early walking impairment, patients quickly lose the ability to move independently.
- Oromandibular. Muscular dystonia in the mouth and lower jaw. Patients experience spasm of the jaw muscles, which makes it difficult to open the mouth voluntarily. In this case, involuntary movements are noted: protruding the tongue, stretching the lips. There may be difficulty swallowing, head tilt, body rotation.
- Loss of central vision, predominance of peripheral vision - patients look at objects sideways.
- Loss of peripheral vision, predominance of central vision (tunnel vision).
- Night blindness.
- Decreased visual acuity.
- Dislike of blinding bright light.
- The result is complete loss of vision.
Features of the clinical picture
Signs of this pathology may vary in each specific case, and the manifestation of symptoms depends on the form of the disease in the individual.
The childhood form of Hallervorden Spatz appears between the ages of 5 and 10 years. It is considered a classic form of the disease.
In the vast majority of cases (90%), the disease begins with torsion dystonia, which affects the leg muscles. The patient experiences muscle contraction, difficulty walking, and changes in gait. Then the muscles of the face, pharynx and trunk are affected.
The presence of blepharospasm, hand spasm, spasmodic torticollis, and facial hemispasm may be present. A third of patients have muscle rigidity, hypokinesia, and tremor (signs of Parkinsonism syndrome).
Typical signs of the childhood form of the disease also include:
- epileptic syndrome;
- aggressiveness;
- mental retardation (occurs due to deterioration of memory and attention);
- asociality;
- visual disturbances (optic atrophy, retinal degeneration);
- mental disorders.
Differential diagnosis
The clinical picture of GSH resembles many other diseases of the nervous system, the main manifestation of which is parkinsonism.
- Wilson-Konovalov disease. Pathology associated with impaired copper metabolism. It is transmitted hereditarily by the same mechanism as BHS. The symptoms are similar: muscle dystonia, dementia, dysarthria, dysphagia. There is a pathognomic symptom - unusual eye color due to Kayser-Fleischer rings. Diseases are distinguished based on the results of MRI and DNA analysis.
- Huntington's disease. A hereditary disease of the nervous system that is associated with atrophy of the striatum (one of the structures of the basal ganglia) and the cerebral cortex. Manifested by erratic movements and decreased cognitive abilities. Unlike BHS, it often appears after 35 years. Diagnosis is based on genetic testing and MRI.
- Farah's disease. Degenerative disease of unknown origin. Like BHS, it affects the basal ganglia, but instead of iron, calcium is deposited in them. MRI and CT show a symmetrical lesion with calcifications. Clinical manifestation is parkinsonism. Symptoms can first appear at any age.
Diagnostic criteria and techniques
Main criteria for diagnosis:
- onset of illness before age 30;
- typical picture in MRI results;
- constant development of symptoms;
- extrapyramidal syndromes;
- pyramid signs;
- epileptic seizures;
- cognitive impairment;
- retinal pigmentary degeneration;
- presence of the disease in a family history.
The disease must be differentiated from other diseases that have a similar clinical picture: Huntington's chorea, Wilson-Konovalov disease, choreoacanthocytosis, Machado-Joseph disease.
For diagnosis, the patient is referred for the following examinations:
Treatment of Hallervorden-Spatz disease
Currently, no effective treatments have been developed for Hallervorden-Spatz disease. In this regard, symptomatic treatment is used. Parkinsonism syndrome is an indication for the use of dopamine agonists or amantadine derivatives. However, with this disease, it is usually resistant to treatment.
For hyperkinesis, the use of valproates and benzodiazepines is indicated. For spastic symptoms, muscle relaxants are recommended, for epileptic seizures - topiramate or valproate, for cognitive disorders - ipidacrine and choline alfoscerate, for mental disorders - antipsychotics.
Treatment of patients with Hallervorden-Spatz disease remains mainly symptomatic, that is, aimed at relieving symptoms rather than eliminating the cause. It is currently not possible to eliminate the genetic abnormality that leads to HSP.
Tremor is effectively treated with dopaminergic agents. The anticholinergic drug benzotropine can be used to alleviate muscle stiffness and tremors, as well as dopamine agonists.
To eliminate dysarthria and drooling, the following are recommended:
- methscopolamine bromide;
- speech therapy manipulations.
Systemic chelating agents such as desferrioxamine have previously been used to remove excess iron from the brain, but their effectiveness has not been proven. Dementia is a progressive symptom and its progression can be slowed, but the condition cannot be cured.
Recommended medications for Hallervorden-Spatz disease:
- baclofen;
- benztropine;
- memantine;
- rivastigmine;
- donepezil.
Recommended activities:
- physiotherapy;
- occupational therapy;
- speech therapy.
There is currently no cure for Hallervorden-Spatz disease. Previously, they tried to influence the accumulation of iron in the cells of the basal ganglia. However, this treatment regimen did not prove effective.
Research is underway on drugs belonging to the group of detoxifying agents. It is assumed that they are able to slow down the progression of HSP. Clinical trials have shown that this is indeed the case, but the difference is not significant.
To alleviate the patient's condition, symptomatic medications are used.
- Treatment of parkinsonism. Dopamine agonists alleviate symptoms, but they are significantly less effective in HSP. Therefore, tranquilizers are more often prescribed for hyperkinesis.
- For muscular dystonia, muscle relaxants are used.
- For epileptic seizures - antiepileptic drugs of the valproate group.
- For signs of severe mental disorders - antidepressants and antipsychotics.
Therapy helps improve the course of the disease and alleviate the main symptoms. However, taking medications does not affect life expectancy in any way and does not improve the prognosis.
https://www.youtube.com/watch?v=https:accounts.google.comServiceLogin
Experimental tools are being developed. These include experimental surgical treatment methods: pallidotomy and thalamotomy. However, most patients who undergo surgery return to the original level of manifestations within a year after surgery.
Generally unfavorable, as the disease is incurable. The timing of the onset of disability depends on the clinical form of the disease. The most favorable prognosis is with a late onset of the disease. In this case, dementia is mild, symptoms progress slowly.
With early manifestation, manifestations are stable and progress quickly. Complete loss of the ability to self-care occurs 10 years after the onset of the disease.
Do not neglect treatment. It improves the patient’s quality of life and makes it possible to delay the moment of disability.
How can modern doctors help?
There are no treatments in modern medicine that can prevent or stop Hallervorden-Spatz disease.
Therapy is aimed at alleviating and relieving the intensity of symptoms:
- For parkinsonism syndrome, dopamine agonists (Pronoran, Pramipexole, Mirapex, Piribedil) and amantadines (Simmetrel, Midantan) are prescribed. However, the syndrome is often resistant to treatment.
- To relieve hyperkinesis, valproates (Konvulex, Depakin, Encorat) and benzodiazepines (Clonazepam, Diazepam) are used.
- To relieve muscle spasticity, muscle relaxants (Mydocalm, Baclofen) are used.
- Epileptic seizures are relieved with valproate and Tomapax.
- For cognitive impairment, Gliatilin and Neuromidin are used.
- For the treatment of mental disorders, it is recommended to take antipsychotics (Clonazepam, Quetiapine, Rispolent), antidepressants (Dapoxetine, Citalopram, Venlafaxine).
New methods of treating the disease are also emerging. These include therapy by administering pantothenic acid, magnetic stimulation of the brain (globus pallidus).
Forecast
Hallervorden-Spatz disease is characterized by a steady progression of symptoms. The most aggressive course is characteristic of the childhood form: 6-15 years after the onset of the pathology, complete disability occurs.
The late version of the disease has a more favorable prognosis, especially if accompanied by mild dementia. Through therapy, the severity of symptoms can be reduced and the patient's ability to self-care can be maintained. The average duration of the atypical form of the disease is 20 years or more.
Literature
- Hochspringen ↑ Zhou ua: A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. in: Nature genetics (Nat Genet.). New York 28.2001,4 (Aug), 345-349. PMID 11479594 ISSN 1061-4036
- Hochspringen ↑ PKAN-Artikel bei genereviews von Gregory/Hayflick, revidierte Version Jan. 2008
- Hochspringen ↑ M. Shevell: Hallervorden and history. in: The New England journal of medicine (N Engl J Med.). Waltham Mass 2.2003,1 (Jan),348,3-4. PMID 12510036 ISSN 0028-4793
This page was last edited on December 26, 2019 at 12:14 pm.
general information
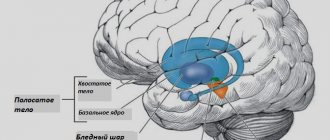
The basal ganglia are neural subcortical ganglia located in the central white matter of the hemispheres. They consist of the striatum, striatum and lenticular nucleus. The structure of the latter includes the globus pallidus and the shell. In addition, part of the substantia nigra, a part of the extrapyramidal system, belongs to the basal ganglia.
Purpose of the basal ganglia:
- regulation of motor and autonomic functions (breathing, cardiovascular system);
- participation in integrative processes of higher nervous activity.
With Hallervorden-Spatz disease, iron accumulates in the globus pallidus and substantia nigra, which leads to disruption of their function. As a result, motor dysfunctions and mental disorders occur. The disease was first described in 1922 by physicians Julius Hallervorden and Hugo Spatz. It occurs in 3 out of 1,000,000 people.
Course of the disease
The usual onset of the disease is in childhood, sometimes with severe symptoms already in the first year of life. Manifestation in adulthood is rarely possible. First, extrapyramidal motor disorders occur, especially gait disturbances with a tendency to fall or leg dystonia, and less commonly, mental disorders.
Subsequently, movement disorders (dystonia, choreoathetosis, tremor) with neck rigidity, hyperreflexia and mental disorders (as a rule, intellectual disorder in the form of progressive dementia). Dysarthria and dysphagia are also found. Retinal pigmentation or optic nerve atrophy may be detected[2].
Death usually occurs before the age of 30, in a state of complete mental collapse of the personality.
Due to Julius Hallervorden's connections to the euthanasia program in Nazi Germany, it was proposed to call the disease "neurodegeneration with brain iron accumulation 1". The abbreviated name of the disease as “NBIA1” is widespread throughout the world.