Children with Angelman syndrome: adaptation and education
“Elf children” or Williams syndrome is a genetic disorder characterized by the absence of several genes in the human genome.
The total number of genes in such patients can range from 25–29. Depending on the number of missing units of genetic material, the symptoms and severity of pathological changes change. In the course of lengthy laboratory studies, geneticists were able to establish that elf children are born as a result of a deletion of the 7th chromosome near its long arm. The length of the missing section can vary between 1.6–1.8 million nucleotide pairs. A disturbance in the composition of the gene apparatus is provoked by a spontaneous mutation.
However, there is evidence indicating an autosomal dominant type of inheritance of the pathology. Compared to other genetic hereditary pathologies, Williams syndrome is a common disease: the incidence can reach 1 case per 20,000 newborns. At the same time, there is no pattern of gender distribution of the disease: girls and boys are affected by the disease with the same frequency.
As the results of genetic studies have shown, elf children (Williams disease) are born as a result of a mutation occurring at the stage of meiosis. Chromosomes are damaged during the formation of sex parent cells. If one of them has an anomaly, there is a high probability of abnormalities developing in the embryo. The absence of the ELN gene on the chromosome causes insufficient elastin formation.
This substance is part of the walls of large blood vessels and heart valves. As a result of such changes, patients with Williams syndrome develop pathologies of the cardiovascular system, heart defects, and arterial hypertension. The absence of the LIMK1, GTF2IRD2 genes causes pathological changes in neurons, which result in neurological abnormalities.
Children with doll syndrome need more attention than healthy babies. A sick child is usually hyperactive, more drawn to people, loves to play and communicate.
Characteristic signs in behavior and communication:
- Excessive friendliness;
- Good nature;
- Attachment to people.
Mental retardation may not be very pronounced - children distinguish emotions well enough and understand others. But at the same time, such children are very gullible and do not understand well when they are being deceived.
The disease does not affect life expectancy. Parents need to help such children adapt, follow a diet and sleep schedule that may cause problems. Most of these children master all self-care skills as they grow up.
General information
The syndrome is named after the British pediatrician G. Angelman. In 1965, he was the first to describe the symptoms of the disease and called it “happy puppet syndrome” because the patients reminded him of the character in the film “The Boy Puppet.” In those years, genetic research methods had not yet been developed, and it was impossible to establish the cause of the pathology.
In 1987, researchers determined the etiology of the disease and renamed it Angelman syndrome. Now this term is official, but you can find synonymous names - “puppet syndrome”, “Parsley syndrome”, “happy doll syndrome”. The prevalence is 1 case per 10-20 thousand newborns. The disease is detected after the first year of life (sometimes by 3-7 years), boys are more often affected.
Angelman syndrome
Angelman syndrome is a pathology that occurs as a result of genetic mutations, in particular due to the loss of a fragment of chromosome 15.
A genetic mutation can manifest itself as a result of violations of chromosomal inheritance, when a child inherits 2 paternal chromosomes, instead of 1 paternal and 1 maternal, when a fragment of a chromosome is moved, or when one or another of its fragments is changed.
The disease is not always inherited; it often happens that a sick child is born to parents who do not suffer from any genetic abnormalities.
Thus, we can conclude that the mutational processes leading to the onset of the disease are spontaneous, and predicting the birth of a child with such abnormalities is very problematic.
How is Elf syndrome treated?
Patients diagnosed with Williams syndrome (Elf face) receive treatment only in the form of symptomatic therapy, since genetic diseases currently have no specific treatment.
For children admitted to the hospital with severe hypercalcemia, glucocorticoids (Hydrocortisone) are prescribed, and adult patients are usually recommended to constantly monitor blood calcium levels and vitamin D intake, which increases calcium levels.
Children with this syndrome require intensive training with a psychologist. Contact with healthy children is also of great importance for such patients, as it creates positive dynamics for them in their overall development.
Clinical characteristics of the child
Different patients may exhibit different symptoms. This difference depends on the degree and type of chromosomal abnormality.
Signs can be divided according to the frequency of their manifestation in children with Angelman syndrome:
- Symptoms that appear in all patients. These include severe functional developmental delay, behavioral deviations (laughter and smiling for no reason, a state of happiness, increased excitability, decreased concentration). Also, absolutely all patients experience impaired motor functions, impaired balance, tremors, a predominance of nonverbal skills over verbal ones, and speech disorders.
- Symptoms characteristic of 80% of patients. Stunted growth leading to disproportion of the head relative to other parts of the body. This often leads to the development of microcephaly. Children under three years of age often experience epileptic seizures, then they become less frequent or disappear altogether. Electroencephalogram results are often abnormal, with low-level waves having increased amplitude and temporal dynamics.
- Symptoms that occur in less than 80% of patients. Such manifestations include strabismus, decreased ability to control tongue movements, problems with swallowing, hypopigmentation of the eyes and skin, albinism. Many patients experience increased activity of tendon reflexes. In early childhood, many people have problems with eating and sleeping. Manifestations also include drooling, tongue protruding, and constant thirst. External signs also include the presence of a flat back of the head and smooth palms.
As you grow older, the manifestations of the syndrome change. Sleep disorders, hyperactivity, and seizures are less common. Adults with Angelman syndrome appear younger than their peers.
Puberty occurs a little later than in individuals who do not have this pathology. Patients can have their own children, but there is a high risk of transmitting the disease to their offspring.
Many adult patients suffer from uncontrollable urination. Often there are difficulties with motor skills, which leads to the need to wear clothes without zippers and buttons. Among adult patients, there is a problem of excess weight, so it is necessary to monitor compliance with a special diet.
Children perceive oral speech well and understand the content of almost all conversations, but rarely respond. They often refuse to take part in the conversation and use several dozen words in their speech.
How to recognize Williams syndrome (“Elf face”)
In addition to the characteristic appearance, patients with Williams syndrome also have features of the development of higher nervous functions, which lead to the emergence of specific features in the psyche.
- Impaired sensory integration (the process by which the nervous system receives signals from the senses of touch, vestibular system, smell, vision, taste and hearing). At the same time, sensitivity to sound in patients is increased.
- Patients are impulsive, easily excitable, very sociable (sometimes even obsessively), their mood is unstable (in medicine, this feature is characterized as emotional lability).
- They experience increased anxiety and a noticeable fear of everything new.
- Characterized by impairments in the ability to pronounce sounds (expressive speech) and perceive what is said (impressive speech).
- During training of such patients, problems with mastering mathematical operations are noticeable.
At the same time, the way mental retardation manifests itself in such children looks unusual. Their speech, as a rule, is well developed for their age, expressive and emotionally colored, in addition, they have obvious musical abilities: absolute pitch, an excellent sense of rhythm and an excellent musical memory.
Diagnostics
This disease can be detected even before the baby is born. For this purpose, invasive or non-invasive diagnostics are used. In the first method, amniotic fluid is taken for research. Invasive diagnostics is considered unsafe, since there is a high risk of developing a uterine infection. In the second research method, Angelman syndrome in children is identified by blood donated by pregnant women. Geneticists analyze the biomaterial, study the structure of the baby’s DNA and determine whether there are deviations or not.
It is important for parents to closely monitor the condition of the baby and, if they detect the first warning signs, immediately seek medical help.
If parsley syndrome is previously diagnosed as a disease, the following study is prescribed to confirm the presence of pathology:
- general examination of the patient;
- psychiatrist consultation;
- neurological examination;
- laboratory diagnostics.
Most often, the diagnosis is made when the child reaches the age of 3-7 years. During this period, symptoms of the disease appear, which are used to preliminarily determine its presence.
During the neonatal period, it is quite difficult to identify pathology, since there are no clinical manifestations and are hidden. In addition to analyzing the signs of the disease.
A number of studies are carried out, such as determining the tone of muscle tissue, as well as genetic research on chromosome 15. Violations of this element indicate the presence of Angelman syndrome.
Neurologists, psychiatrists and geneticists examine children with suspected Angelman syndrome. Parents complain of lack of speech, motor stereotypies, difficulties in developing walking and other motor skills, and hyperactivity. A differential diagnosis is carried out, during which more common diseases should be excluded, such as mental retardation, autism spectrum disorders, dementia, mutism, and non-symptomatic forms of epilepsy. A comprehensive study includes the following procedures:
- General inspection. The presence of the syndrome is often indicated by specific features of the patients' appearance: protruding tongue, drooling, large mouth, wide sparse teeth, protruding lower jaw, flat shape of the back of the head. Characterized by a light shade of skin, eyes and hair. The gait of children resembles the movements of a marionette doll due to increased tendon reflexes and decreased muscle tone.
- Examination by a psychiatrist. In 100% of cases of Angelman syndrome, a pronounced delay in mental development, lack of independent speech, or a very poor vocabulary is diagnosed. Communication is carried out using facial expressions, gestures, and drawings. Behavior is marked by hyperactivity, stereotypical hand movements, and causeless laughter.
- Neurological examination. All patients have ataxia and tremor of the limbs. 80% of patients have postnatal microcephaly - the newborn’s head circumference is less than 32 cm, by 12 months - about 42 cm. EEG data reveals symptomatic epilepsy (high-amplitude discharges of slow complex waves), convulsive seizures are clinically possible. Some children have strabismus, a diffuse decrease in muscle tone, increased tendon reflexes, and hyperkinesis.
- Genetic research. Laboratory diagnostics are aimed at identifying mutations and deletions in the UBE3A gene. A set of procedures is sequentially carried out, including fluorescence in situ hybridization, analysis of the imprinting center mutation, DNA methylation of the CA/PVS region, diagnosis of deletion using microarray analysis, and search for mutational changes in the UBE3A locus.
Treatment of Angelman's disease
Therapy for Angelman syndrome is symptomatic and supportive. Several clinical trials are ongoing, but there are no genetic therapies or cures available yet. The general physical health of children with happy doll syndrome is good. Routine pediatric care and routine immunization are indicated.
Anticonvulsant
Patients with Angelman syndrome who experience seizures need anti-seizure (anticonvulsant) medications. Seizures are usually adequately controlled with one medication. In some cases, control is difficult and multiple medications are needed.
No single anticonvulsant drug has been shown to be most effective in all cases. Sleep disorders are common and require strict sleep rituals and behavioral therapy. Sometimes the use of sedative medications is required.
Feeding problems in newborns are addressed through modified breastfeeding techniques or aids such as special nipples to help babies with poor sucking ability.
Gastroesophageal reflux is treated with upright positioning and medications to help move food through the digestive system (motility medications).
In some cases, surgical tightening of the valve that connects the esophagus to the stomach (esophageal sphincter) may be necessary. Laxatives are used to treat constipation.
Musculoskeletal
Ankle braces/supports and physical therapy help learn to walk. Scoliosis develops in about 10% of cases and requires braces or surgical correction. Strabismus is often corrected surgically.
Early intervention is important to ensure that children with Angelman syndrome reach their potential. Special services include specialist support, medical and psychological assistance. Most children receive physical, speech and psychological therapy. Corrective behavioral therapy is used to prevent unwanted behavior.
Families with Angelman syndrome will benefit from genetic counseling.
There is no special therapy. Drug treatment for seizures is usually necessary. Physical, communication, and behavioral therapies allow people to achieve their maximum development potential.
The chromosomal abnormalities underlying the syndrome cannot be eliminated. Patients are prescribed symptomatic treatment, psychological and pedagogical correction, and rehabilitation measures. Anticonvulsants are used to reduce the frequency of epileptic seizures, and melatonin is used to normalize sleep. Therapeutic physical education classes and massage sessions are aimed at developing fine motor skills and coordinated gait, eliminating scoliosis. To improve communication skills, children are taught sign language, involved in group activities, and behavioral therapy sessions are organized to help them learn the rules of interaction in society.
The search for ways to effectively treat the syndrome continues. The drugs are being tested on genetically modified mice. The results prove that topoisomerase inhibitors are able to activate the maternal UBE3A gene. At this stage, control studies are carried out, the safety and risks of therapy are determined, but there is not yet enough information to transfer experiments to groups of people.
To date, a miracle drug has not yet been invented that would help defeat this genetic disease. However, Angelman's disease requires symptomatic therapy, which alleviates the patient's condition. In this case, drug and non-drug treatment is prescribed. Parsley syndrome in children requires the following therapy:
- Anticonvulsants are prescribed. Clonazepam, Convulex, Lamotrigine are most often prescribed. Such medications minimize the frequency and intensity of epileptic attacks.
- Vitamin therapy is prescribed (elements of groups B, C, D and E). This treatment strengthens the body's immune system. However, it reduces the effectiveness of antiepileptic drugs, so all prescriptions must be made by a doctor.
- Therapy is prescribed to eliminate problems with the digestive tract. This treatment involves taking laxatives (Fitolax or Senade) and probiotics (Hilak forte, Bifiform).
- Sleeping pills are prescribed. Most often, patients are prescribed Diphenhydramine or Melatonin.
- Hormone therapy is indicated. This treatment is aimed at correcting the behavior of patients. For example, the hormone secretin improves the digestion process, and oxytocin increases cognitive abilities and memory.
- Physiotherapy will help in the fight against joint problems. Paraffin baths, massage, magnetic therapy, electrophoresis, and aqua gymnastics can be prescribed.
For this disease, behavioral therapy, classes with a speech therapist, psychologist and speech pathologist are indicated. Children diagnosed with happy doll syndrome have severe speech impairments. Some of them have a limited vocabulary, and others even have difficulty producing individual sounds.
Classes with such children should be short (no more than 30 minutes), but regular: it is advisable that they be held every day. According to experts, you can start with finger painting. During such activities, the child calms down, gets acquainted with the color scheme and develops fine motor skills.
Since children with Angelman syndrome perceive information better when it is visualized, it is advisable to use flashcards and other visual aids when teaching them. For example, to explain to such a child the concepts of “big” and “small”, you can use a collapsible nesting doll. When a child first encounters such a visual aid, the adult must open the toy himself and comment on each of his actions. Then you can ask your child to do the same. Gradually the task becomes more complicated: for example, the baby must show where the big matryoshka is.
Russian Medical Server / Treatment in Italy / Center for the treatment of rare diseases in Milan / Angelman syndrome - treatment in Italy
Angelman syndrome is a fairly rare disease caused by chromosomal abnormalities and occurs in approximately 1 in 10,000 newborns. Children with this defect experience delayed mental and neurological development from the first years of life.
Since we are talking about a genetic defect, there is no drug treatment for it, however, there are procedures that can improve the quality of life of patients diagnosed with Angelman syndrome. For example, from early childhood, experts recommend giving the patient massage and physical procedures to strengthen the body.
With proper care, people with this disorder can live into adulthood and care for themselves, although they may never be completely independent.
The disease is named after Harry Angelman, a pediatrician from Great Britain who first diagnosed the above-mentioned disorder in 1965, calling patients “doll children.”
As a doctor at Warrington Hospital, the medic cared for three young disabled patients with different symptoms that initially seemed to be the result of different diseases. However, the specialist felt a commonality between them. Based on clinical studies, Angelman made a general diagnosis, but could not prove it at a more precise level, since the laboratory equipment of that time did not allow such complex genetic studies. For this reason, Angelman was in no hurry to publish his thoughts in medical journals.
The pediatrician was given confidence in these guesses by the painting “Boy Puppet”, painted in oil, which he saw in the Italian museum in the city of Verona. The picture shows a laughing boy. He reminded the doctor of all three of his patients so much that Angelman decided to write one general article about them, called “Children Puppets.”
The scientific work was published in 1965. She aroused a little interest, but was soon quickly forgotten about. Doctors return to the article already in the eighties, when references to the disease began to appear in the medical community. Only by 1987, scientists were able to establish a pattern that most children with the syndrome are missing part of chromosome 15.
Due to the fact that the name of the diagnosis “happy puppet syndrome” seemed scary and humiliating to many parents, it was later renamed using the name of the scientist Angelman.
The syndrome is diagnosed by genetic analysis (chromosome 15), recommended for newborns with reduced muscle tone (hypotonicity), delays in the development of gross motor skills and speech development. Parents and doctors should pay attention to cases of small tremors, chaotic, jerky movements of the limbs, gait with stiff legs; in some cases, a specific facial expression, too frequent laughter.
Possible analysis methods: fluorescence in situ hybridization process, DNA methylation in the 15q11-q13 region, imprinting center mutation analysis, direct mutation analysis of the UBE3A gene.
Genetic analysis allows us to identify a number of pathologies that may be detected in an unborn child.
Chromosomal diseases are quite severe, and treatments for them have not yet been found. For this reason, parents must prepare for what the future holds. In some cases, genetic testing for chromosomal diseases is necessary in order to decide whether to terminate a pregnancy.
Angelman syndrome is a congenital genetic abnormality; Currently, specific methods for its treatment have not been developed, but some therapeutic measures improve the quality of life of people with the syndrome. Infants with hypotonia should receive massage and other special therapies (physical therapy).
Symptoms
The first signs of a pathological disorder begin to appear at the age of 6 months. As the baby grows, they become more distinct. Parsley syndrome symptoms are as follows:
- Group of physical signs. Patients diagnosed with this condition have vision problems. More often it is strabismus, the presence of spots on the iris. In addition, in such patients the proportionality of the body is disturbed: the head is small compared to other parts of the body. At the age of 12 months, another accompanying sign begins to appear - the baby opens his mouth wide and slightly sticks out his tongue. Some of these patients exhibit hypopigmentation of certain areas of the skin.
- Neurological signs. Patients experience sleep disorders. At the same time, disturbances in speech functions are noted. A child aged 3 years can often suffer from epileptic seizures. However, the severity and intensity of such signs decrease as the child grows older.
- Psychological signs. Patients have mental retardation and affective behavior.
This disease has its own clinical picture. Parsley syndrome is a disease accompanied by the following symptoms:
- severe developmental delay;
- tremor;
- imbalance;
- increased thirst;
- increased chewing movements;
- flat palms;
- problems with fine motor skills (difficulty fastening buttons and zippers);
- uncontrolled urination;
- excess weight;
- pointed chin;
- the lower part of the jaw protrudes slightly forward;
- rachiocampsis.
This disease is characterized by:
- laughter and joy for no reason;
- chaotic gesturing;
- difficult or absent speech;
- imbalance;
- slow weight gain;
- seizures;
- sleep disturbance;
- strabismus (squint);
- convulsions;
- excessive salivation;
- tongue prolapse;
- chronic thirst.
Symptoms in adult patients soften over the years. By adulthood, the manifestation of some symptoms, such as hyperactivity and sleep disturbances, weakens, while some, on the contrary, may worsen, for example, scoliosis. As you age, weight problems may begin. Girls with this syndrome may experience an increase in seizures during puberty, which occurs at normal times.
The first signs of parsley syndrome appear in the early months of development.
- Problems with feeding, sleeping, slowing down weight gain. The need for sleep may decrease, and normal sleep patterns may not be established for a long time.
- At school age, there will be problems with reading and perception, difficulties with concentrating.
- Hyperactivity, speech problems and poor coordination can interfere with learning.
- Because of their stiff-legged gait, patients are called “marionettes,” they are also characterized by scoliosis and, worst of all, epilepsy is observed in 80% of patients.
Clinically, the disease manifests itself between the ages of 6 and 12 months. The developmental delay gradually increases; previously mastered skills are retained, but the acquisition of new ones occurs slowly. Children can sit, crawl, pick up objects and move them from hand to hand, maintain eye contact, walk and babble. Walking is difficult, the sense of balance is impaired, frequent falls and bruises on furniture are observed. Tremors and chaotic movements of the limbs, especially the hands, are detected. Speech disorders include both delays and a complete lack of expressive speech. Children either do not speak at all or use babbling, simple syllables and words with a total volume of no more than 10 units. The understanding of spoken speech, the desire to communicate, and the use of nonverbal means of communication: gestures, facial expressions, and indirect signs are preserved.
The main behavioral disorder is hyperactivity. Children often have fun and laugh without an objective reason; they are motorically disinhibited, restless, and unfocused. Many people develop a pathological attachment to a certain toy or household item, the appearance of which immediately increases the mood, capriciousness and crying are replaced by laughter. Concentration is reduced, switching is rapid and undirected. There are learning difficulties and a persistent decline in intellectual functions. Stereotypes easily become established: body swaying, arms waving. 80% of patients have microcephaly, insufficient cranial volume, and epileptic brain activity. Rarely, there is a decrease in control of tongue movements, which is manifested by difficulty sucking the breast or pacifier, followed by lack of body weight.
Characteristic features of the appearance of children are strabismus, scoliotic curvature of the spine, enlarged teeth and lips, thinning of the dentition, flattening of the back of the head, protrusion of the chin. The tongue is often stuck out, the mouth is slightly open in a smile. Muscle dystonia develops, the severity of tendon reflexes increases, forming the specificity of motor skills: patients walk on straight, unbending legs, raise their shoulders, and bend their arms at the elbows. A peculiar symptom is a craving for water. Most children feel calmer in an aquatic environment; they like to splash around in the bathtub and play with boats in the basin.
The clinical picture of the disease is very diverse and may differ in each specific case. In particular, the child exhibits physical and mental abnormalities, some signs are characteristic of all cases, others are much less common.
| Physical abnormalities | Psychical deviations |
|
|
Clinical manifestations of the disease are also usually divided depending on their prevalence. Thus, there are symptoms that occur in all children suffering from this disease, signs that occur in most cases (but not always) and more rare manifestations.
| Characteristic symptoms | Common signs | More rare manifestations |
|
|
|
The clinical manifestations of the disease change as the child grows older. Thus, in adolescent children there is a delay in puberty (while childbearing function is not impaired in the future), and when the child becomes an adult, he looks much younger than his peers.
Often, older children experience other manifestations of the disease, such as difficulty urinating, complex motor skills of the hands, problems with communication (the teenager understands spoken language, but does not see the need to respond to the interlocutor).
Outwardly, a sick child looks quite content and happy, however, this impression is deceptive.
Such children always have a severe delay in mental and physical indicators, which leaves a negative imprint on learning and communication with peers.
In severe cases, a persistent and significant impairment of hand motor skills develops, which makes the child unable to take care of himself (in particular, the child cannot independently fasten a button or zipper on clothes or shoes, or perform any other actions that seem quite ordinary to a healthy person) .
All this significantly worsens the child’s quality of life.
Children suffering from Angelman syndrome also have external differences, such as:
- flat face (its middle part), insufficient development of the facial bones of the skull;
- small and pointed chin;
- hypertrophied lower jaw, which protrudes strongly forward;
- a large and wide tongue that the child often sticks out;
- violations of the dentition, the presence of interdental spaces;
- small skull (the rest of the bones of the skeleton are of normal size, corresponding to age indicators);
- deformation (curvature) of the spine.
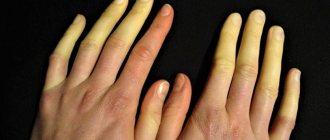
The main visual symptoms of the disease, which are determined in infancy, include specific facial features and the structure of the skull. Thanks to this feature, the anomaly received its second name - “elf face” syndrome. As the child develops, the skeleton and muscle structure changes. Characteristic appearance features:
- high, wide forehead, disproportionate to the small chin, narrowed downwards;
- the flat bridge of the nose goes into a short nose, which has no tip;
- the section of the wide-set eyes is narrow, the color of the pupil is blue, the eyelids are swollen, convergent squint;
- low-set ears elongated upward, which makes the individual look like a fantasy character;
- the mouth is large, the upper lip is much thinner than the lower.
The doll-like appearance is complemented by plump, drooping cheeks and straight, well-shaped eyebrows.
In children with this diagnosis, teeth erupt later than normal, the jaw becomes deformed, and signs of malocclusion appear. Lack of appetite affects growth, and weight loss is noted. Muscles are underdeveloped. Secondary symptoms of the syndrome include flat feet, weak vestibular apparatus, and poor coordination.
Complications
Angelman syndrome is a rare, little-known disease. In this regard, the diagnosis and provision of medical, psychological and pedagogical assistance are often carried out untimely, by 6-8 years, which causes low success of correctional and therapeutic measures. Without physiotherapeutic treatment, disorders of the musculoskeletal system worsen - patients suffer from severe forms of scoliosis and have difficulty moving independently.
Angelman syndrome is a rare, little-known disease. In this regard, the diagnosis and provision of medical, psychological and pedagogical assistance are often carried out untimely, by 6-8 years, which causes low success of correctional and therapeutic measures. Without physiotherapeutic treatment, disorders of the musculoskeletal system worsen - patients suffer from severe forms of scoliosis and have difficulty moving independently.
Causes
The main cause of the development of the disease is considered to be a change or absence of one of the fragments of chromosome 15. The risk of developing pathology in a child increases if his close relatives (parents) have the following genetic abnormalities:
- The presence of extra chromosomes (one or more).
- Chromosomal inversion, when one of the sections of the chromosome is turned upside down, while part of its fragment is lost, and the genes are located in the opposite order.
- Rearrangement of the Y chromosome, when 2 sections of it change places, while some fragments of the mutated chromosome may be missing.
- Loss of one or more chromosomal fragments.
- Attachment of a fragment of one chromosome to another.
- The presence of extra genes resulting from the replication of one or more chromosomes.
- The ring structure of a chromosome is when its ends are connected to each other, forming a ring.
Factors in the development of Angelman syndrome continue to be studied. A genetic defect was found on chromosome 15 of the maternal set, but its nature and method of occurrence may vary. Sometimes the disease debuts as a result of the transfer of altered genetic information from a parent, sometimes it is a consequence of spontaneous disorders in the genome. Chromosomal abnormalities can be detected in approximately 85-88% of patients. The cause of the syndrome may be:
- Deletion. With this defect, part of the genetic material is lost or inactivated. In 70% of patients, extensive deletions of the 15q12 region of chromosome, in which the gene activator is localized, are diagnosed.
- Uniparental disomy. ARF is determined in 2-3% of cases of the disease. The chromosome set contains two copies of the father's chromosome 15. There is no maternal chromosome, and the gene is also missing.
- Imprinting defect. The essence of the anomaly is that the imprinting center, which regulates the activity of the UBE3A locus, turns out to be non-functional, “turned off”. The gene remains structurally intact, but does not perform its functions. The prevalence of ID is 3-5%.
- UBE3A mutation. In 5-10% of patients, the cause of the disease is mutational changes in the gene. They are represented by inversions, microdeletions, translocations and duplications.
History of the study of the disease
The disease is named after Harry Angelman, a British pediatrician who first differentiated this syndrome in 1965. Then it was called happy puppet syndrome, but today this term is not used, as it was considered disparaging.
Dr. Angelman had treated several children with similar symptoms and suspected a common diagnosis. It was not possible to prove the diagnosis and obtain accurate data at that time due to the lack of technologies that are available today. The doctor reflected his guesses in an article entitled “Children of Puppets.”
At that time, the publication did not arouse much public interest, and was soon forgotten. It was remembered again in the eighties, when the necessary research methods appeared. Scientists have found that most children suffering from the syndrome are missing a small part of chromosome 15.
Risks
Assessing the risk of having a child with Angelman syndrome again in the same parents is very difficult; consultation with a professional geneticist is necessary. A common deletion is considered to be spontaneous, with a risk of recurrence of less than 1%. In the case of a molecular microdeletion in 15q11-q13, if it is also observed in the mother, the risk is theoretically up to 50%. Mutations within the UBE3A gene can be random and non-inherited, in which case the risk of recurrence is <1%; however, these mutations can be inherited from a normal mother, giving a theoretical risk of 50%. Parthenogenetic disomy of the 15th pair is a random situation; risk of recurrence <1%. There are several people with AS with unusual transformations on chromosome 15, including the 15q11-q13 region; in these cases, the assessment of the risk of recurrence depends on the chromosomal abnormalities in the parents.
Peculiarities
Angelman syndrome is characterized by:
- In 75% of cases there are problems with nutrition, especially with breastfeeding, such babies do not gain weight well;
- delay in the development of gross motor skills (ability to sit, walk);
- delayed speech development, undeveloped speech (in all children);
- children understand more than they can say or express;
- attention deficit and hyperactivity;
- learning difficulties;
- epilepsy (80% of cases), disorders are also detected by electroencephalography; It is believed that children with Angelman syndrome have secondary (symptomatic) epilepsy;
- unusual movements (fine tremors, chaotic movements of limbs);
- frequent laughter for no reason;
- walking on stiff legs - because of this feature, children with this syndrome have sometimes been compared to puppets;
- head size is smaller than average, often with a flattened occiput;
- sometimes characteristic facial features - a wide mouth, sparsely spaced teeth, a protruding chin, a protruding tongue;
- sleep disorders;
- strabismus (squint) in 40% of cases;
- scoliosis (curvature of the spine) in 10% of cases;
- increased sensitivity to high temperature;
- feel more comfortable in the water.