Агенезия мозолистого тела у плода представляет собой врождённое заболевание головного мозга, которое формируется под действием генетически обусловленных факторов. Данное заболевание является внутриутробной патологией и встречается в очень редких случаях.
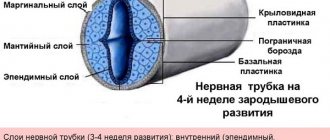
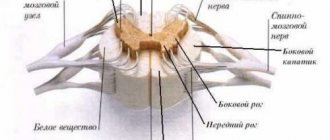
Мозолистое тело образуют нервные окончания, которые переплетают друг с другом. Функцией данного анатомического образования является соединение левого и правого полушария головного мозга. По своей форме мозолистое тело является широким и плоским, а располагается оно под корковым веществом мозга.
Формирование агенезии наблюдается при полном отсутствии спаек, которые соединяют оба полушария. С точки зрения частоты встречаемости, на агенезию мозолистого тела приходится не более 1 случая на 2000 новорожденных детей.
Причины патологии
Аномальное формирование мозга – всегда врожденная патология, появляется она достаточно редко. С морфологической точки зрения сбой происходит примерно на 10-20 неделе беременности, когда начинают закладываться соединительные ткани.
Сила выраженности и проявления может отличаться: спайки либо отсутствуют полностью, либо обнаруживается частичная агенезия мозолистого тела. Факторы, которые располагают к развитию этого состояния:
- изменения в хромосомах, в том числе наследственные;
- спонтанные мутации, не имеющие объяснимых причин;
- внутриутробное инфицирование в результате вирусного заражения матери;
- действие наркотических веществ и препаратов;
- алкогольный синдром у эмбриона из-за злоупотребления матерью;
- нарушения обмена веществ у женщины или дефицит питательных элементов у эмбриона.
Заболевание подлежит внутриутробной диагностике, но точность определения агенезии мозолистого тела низкая.
Лечение и профилактика
Если единственная здоровая почка функционирует в нормальном режиме, симптоматика отсутствует и наблюдается характерное увеличение органа, то врачи не будут применять каких-либо дополнительных действий с целью улучшения работы мочевыделительной системы. Нефролог назначит только динамическое наблюдение за способностью почки вырабатывать и выводить мочу, регулярный анализ мочи и ультразвуковое исследование.
Также предусмотрены профилактические действия, которые направлены на сохранение функциональности почки:
- диета, способствующая снизить нагрузку на единственный здоровый орган, назначается врачом, с учетом индивидуальных особенноcтей пациента;
- ограничение, связанные с контактными видами спорта (бокс, борьба), чтобы обезопасить здоровый орган от травм и лишних повреждений;
- стараться не поднимать тяжелые предметы;
- удерживать постоянную массу тела, избегать резкого похудения;
- отказаться от злоупотребления алкогольными напитками и от никотиновой зависимости;
- поддерживать иммунную систему витаминами и минералами;
- исключить переохлаждения, во избежание возникновения инфекционных процессов.
При отсутствии функции второй почки или при тяжелом развитии агенезии, когда здоровый орган не справляется со своей работой, происходит интоксикация продуктами распада и токсичными веществами. В данном случае назначается гемодиализ и трансплантация почки.
Клинические симптомы
В детском возрасте выявляются агенезия мозолистого тела проявляется только в случае тяжелой формы, так как она сопровождаются выраженной симптоматикой. У взрослых агенезия мозолистого тела находится случайно, в основном при обследовании на другие болезни. Часто такая патология носит легкую форму и не доставляет никаких сложностей в жизни.
У некоторых младенцев заболевание обнаруживается к 3-4 месяцам жизни, когда начинаются проблемы в развитии.
Агенезия мозолистого тела проявляется такими первыми признаками, как: легкие эпилептические приступы, припадки и инфантильные спазмы. Другие симптомы, часть которых можно обнаружить при внутриутробном исследовании:
- изменение процесса формирования мозолистого тела;
- гидроцефалия или микроэнцефалия;
- порэнцефалия, заболевания мантии головного органа;
- атрофия слуховых или зрительных нервов;
- синдром изменения тканей позвоночника, Айкардии;
- кисты и другие новообразования в полушариях;
- плохо сформированные извилины;
- возникновение липом;
- раннее половое развитие в старшем возрасте;
- аномалии внутренних органов;
- часто возникают опухолевые процессы в кишечнике и нарушения в функционировании ЖКТ без видимых причин;
- медленное психомоторное развитие;
- проблемы соматического характера;
- ухудшение координации, слабые мышцы, аномалии скелета.
Агенезия мозолистого тела у плода не проявляется никакими внешними признаками.
Агенезия мозолистого тела
Агенезия мозолистого тела (АМТ) является одним из нескольких нарушений мозолистого тела, структуры, которая соединяет два полушария (левое и правое) головного мозга. При АМТ мозолистое тело частично или полностью отсутствует.
Расстройство вызвано нарушением миграции клеток головного мозга во время развития плода. АМТможет возникать как изолированное состояние или в сочетании с другими церебральными нарушениями, включая порок развития Арнольда-Киари, синдром Денди-Уокера, шизэнцефалию (расщелины или глубокие отделы в тканях головного мозга) и голопрозэнцефалию (неспособность переднего мозга делиться на доли).
У девочек может быть гендерно-специфическое состояние, называемое синдромом Айкарди, которое вызывает серьезные когнитивные нарушения и задержки в развитии, эпилепсия, аномалии в позвоночнике позвоночника и поражения сетчатки глаза.
АМТ также может быть связан с пороками развития в других частях тела, такими как срединные лицевые дефекты. Последствия расстройства варьируются от тонких или легких до тяжелых, в зависимости от связанных с отклонениями в головном мозге. Дети с наиболее тяжелыми пороками развития головного мозга могут иметь нарушения интеллекта, судороги, гидроцефалию и спастичность.
Другие нарушения мозолистого тела включают дисгенез, при котором мозолистое тело развивается неправильно или неполноценно, и гипоплазия, при которой мозолистое тело тоньше, чем обычно.
Люди с этими расстройствами имеют более высокий риск нарушения слуха и сердечных нарушений, чем люди с нормальной структурой.
Нарушения в социальном взаимодействии и общении у людей с расстройством мозолистого тела могут совпадать с поведением расстройств аутистического спектра.
Причины агенезии мозолистого тела
В большинстве случаев причина возникновения агенезии мозолистого тела неизвестна. Однако агенезия мозолистого тела может быть унаследована как аутосомно-рецессивный признак или доминантный признак с Х-сцеплением. Это расстройство также может быть отчасти связано с инфекцией во время беременности (внутриутробной), что приводит к аномальному развитию мозга плода.
Генетические заболевания определяются сочетанием генов для определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Рецессивные генетические нарушения возникают, когда человек наследует один и тот же аномальный ген по одному признаку от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно не проявлять ее симптомы.
Риск того, что два родителя-носителя передадут оба дефектных гена и, следовательно, будут иметь больного ребенка, составляет 25% с каждой беременностью. Риск рождения ребенка, который будет носителем, как и родители, составляет 50% с каждой беременностью.
Шанс на то, что ребенок получит от обоих родителей нормальные гены и будет генетически нормальным для данной особенности, составляет 25%. Риск одинаков для мужчин и женщин.
При Х-сцепленных доминантных расстройствах у женщины будет развиваться болезнь только с одной Х-хромосомой с аномальным геном. Однако у больного мужчины всегда бывает более тяжелое состояние.
Иногда больные мужчины умирают до рождения, так что выживают только женщины. И то только при одной из форм агенезии мозолистого тела, известного как синдром Айкарди. Большинство пациентов, диагностированных до настоящего времени, были женщины.
Синдром Айкарди иногда наблюдается у мужчин с дополнительной Х-хромосомой.
Признаки и симптомы
Агенезия мозолистого тела (АМТ) может первоначально стать очевидным после начало эпилептических припадков в течение первых недель жизни или в течение первых двух лет. Однако не у всех людей с АМТ наблюдаются судороги.
Другими симптомами, которые могут начаться в раннем возрасте, являются проблемы с питанием и задержки в удержании головы в вертикальном положении. Возможны также задержки при сидении, стоянии и ходьбе. Нарушение умственного и физического развития и/или накопление жидкости в черепе (гидроцефалия) также являются симптомами раннего типа этого расстройства.
С помощью неврологического обследования пациентов с АМТ можно обнаружить:
- непрогрессирующую умственную отсталость;
- нарушение координации рук и глаз;
- ухудшение зрительной или слуховой памяти.
В некоторых легких случаях симптомы могут не проявляться в течение многих лет. Пожилые пациенты обычно диагностируются во время тестов на наличие таких симптомов, как эпилепсия, монотонная или повторяющаяся речь или головные боли. В легких случаях расстройство может быть упущено из-за отсутствия явных симптомов в детстве.
У некоторых пациентов могут быть глубоко посаженные глаза и выпуклый лоб. Может присутствовать ненормально маленькая голова (микроцефалия) или иногда необычно большая голова (макроцефалия).
В других случаях АМТ наблюдаються:
- широко расставленные глаза (телеантус);
- маленький нос с перевернутыми (антевертированными) ноздрями;
- аномальные уши;
- чрезмерная кожа на шеи;
- короткие руки;
- пониженный мышечный тонус (гипотония);
- аномалии гортани;
- пороки сердца.
Синдром Айкарди, который, как считается, наследуется как доминантное расстройство, связанное с Х-хромосомой, состоит из агенезии мозолистого тела, инфантильных спазмов и аномальной структуры глаза.
Это расстройство является чрезвычайно редким врожденным расстройством, при котором наблюдаются частые приступы эпилепсии, аномалии среднего слоя глаза (сосудистой оболочки) и слоев сетчатки, отсутствие структуры, связывающей два полушария головного мозга (мозолистое тело), сопровождающая тяжелой умственной отсталостью. Синдром Айкарди в основном наблюдается у женщины.
Синдром Андерманна, выявленный в 1972 году, представляет собой генетическое заболевание, характеризующееся сочетанием агенезии мозолистого тела, умственной отсталостью и прогрессирующими сенсомоторными нарушениями нервной системы (нейропатиями). Ген, вызывающий эту редкую форму агенезии мозолистого тела, был недавно идентифицирован, и в настоящее время доступно генетическое тестирование этого гена (SLC12A6).
X-связанная лиссэнцефалия с неоднозначными гениталиями — это редкое генетическое заболевание, при котором у мужчин маленький и гладкий мозг (лиссэнцефалия), маленький пенис, тяжелая умственная отсталость и трудноизлечимая эпилепсия. Она вызвана мутациями в гене ARX. У женщин те же самые мутации могут вызывать только АМТ, тогда как менее тяжелые мутации у мужчин могут вызывать умственную отсталость. Генетическое тестирование на это расстройство также доступно.
Лечение агенезии мозолистого тела
Лечение симптоматическое и поддерживающее. Противосудорожные препараты, специальное образование, физиотерапия и связанные с ними процедуры могут быть полезны в зависимости от диапазона и серьезности симптомов.
При наличии гидроцефалии ее можно лечить хирургическим шунтом, чтобы слить жидкость из полости мозга, тем самым снижая повышенное давление на мозг.
Генетическое консультирование также может быть полезным для семей с этим расстройством.
Прогноз и осложнения при агенезии мозолистого тела
Прогноз зависит от степени и тяжести пороков развития. Интеллектуальные нарушения не ухудшаются.
Люди с расстройством мозолистого тела, как правило, имеют задержки в достижении основных этапов развития, таких как ходьба, разговор или чтение; проблемы с социальным взаимодействием; неуклюжесть и плохая координация движений, особенно в отношении навыков, требующих координации левой и правой рук и ног (таких как плавание, езда на велосипеде и вождение автомобиля), а также проблемы умственного и социального восприятия, которые становятся более очевидными с возрастом, причем проблемы особенно проявляются в младших классах школа во взрослую жизнь.
Источник: //tvojajbolit.ru/nevrologiya/ageneziya_mozolistogo_tela/
Диагностика
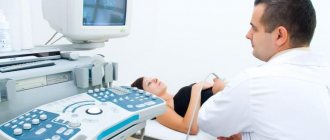
Для обнаружения чаще всего используют УЗИ. Специалист с достаточным опытом и знаниям безошибочно определяет, есть ли у ребенка агенезия мозолистого тела. Дополнительные методики используются, если аномалия носит частичный и слабовыраженный характер.
Дополнительно пациентам назначают МРТ, КТ головного мозга. Иногда требуется сдача сопутствующих анализов крови. Спутать агенезию с другими заболеваниями очень сложно, но для подстраховки некоторые специалисты советуют обращаться за консультацией к 2 и более врачам, чтобы получить более полную картину мнений.
Позволю себе процитировать Марио с недавнего форума по туберозному склерозу.
Normal myelination After normal myelination in-utero, myelination of the neonatal brain is far from complete. The first myelination is seen as early as the 16th week of gestation, in the column of Burdach, but only really takes off from 24th week1.It does not reach maturity until 2 years or so. It correlates very closely to developmental milestones 3. The progression of myelination is predictable and abides by a few simple general rules; myelination progresses from: central to peripheral caudal to rostral dorsal to ventral Myelination milestones term birth : brainstem, cerebellum, posterior limb of the internal capsule 2 months : anterior limb of the internal capsule 3 months : splenium of the corpus callosum 6 months : genu of the corpus callosum Radiographic features CT
Unmyelinated white matter is hypodense compared to normal white matter and grey matter. MRI T1 weighted images is the most sensitive in children less than 1 year of age 1. T2 weighted images is the most sensitive in children between the age of 1 and 2 demonstrating gradual shift from hyer- to hypo-intense relative to grey matter. The only area to remain hyperintense after the age of 2 years, and often for quite some time, is the peritrigonal region 4 FLAIR unsurprisingly follows the same pattern as T2 but lags behind somewhat. The exception is deep cerebral white matter, which begin as heterogenously hypointense during the first few months of life. This area then joins the remainder of white matter as hypointense before finally once more becoming hypointense in the second year of life 2 Proton density weighted images are useful in distinguishing gliosis from hypomylination.
In the acute setting DWI is more sensitive than either T1 or T2.
Fractional anisotropy (FA) increases with brain maturation (diffusion is restriced perpendicular to the direction of axons — thus the increase in DWI signal in large axon bundles running through the slice — e.g posterior limb of interal capsule)
MRS demonstrates elevated myo-Inositol and Choline in neonates. NAA increases with myelination (in the first year of life).
Myelination pattern on MR imaging
Myelination of the brain during infancy progresses in an orderly and predictable fashion.
At birth only certain structures are myelinated dorsal brainstem ventrolateral thalamus lentiform nuclei central corticospinal tracts posterior portion of posterior of internal capsule
Subsequently different parts become myelinated, the first change is increase in T1 signal, and later decrease in T2 2 — 3 months : anterior limb of internal capsule becomes T1 bright 3 months : cerebellar white matter tracts becomes T1 bright 3 — 6 months : splenium of corpus callosum becomes T2 dark 6 months : genu of corpus callosum becomes T1 bright 8 months : subcortical white matter becomes T1 bright 8 months : genu of corpus callosum becomes T2 dark 11 months : anterior limb of internal capsule becomes T2 dark 1 year 2 months : occipital white matter becomes T2 dark 1 year 4 months : frontal white matter becomes T2 dark 1 1/2 years : majority of white matter becomes T2 dark (except terminal myelination zones adjacent to frontal horns and periatrial regions) 2 years : almost all of white matter becomes T2 dark
Лечится ли патология — видео
Вылечить агенезию современная медицина не может, но существуют методики коррекции заболевания. Они подбираются отдельно для каждого клинического случая. Врач учитывает общее самочувствие ребенка, а также выраженность агенезии мозолистого тела.
Для облегчения симптоматики используются специальные лекарства. Однако врачи уверены: большинство методик не способно навсегда избавить от симптомов. Для терапии применяются сильнодействующие средства:
- Фенобарбитал. Уменьшает количество приступов при инфантильных спазмах.
- Бензидиазепины. Психоактивные продукты, которые затормаживают психомоторные реакции и сокращают количество судорожных приступов.
- Кортикостероиды. Необходимы для борьбы с эпилептическими припадками.
- Нейролептики. Используются для лечения психических отклонений.
- Ноотропы. Применяются для коррекции функций головного мозга.
- Диазепам. Корректирует поведенческие расстройства.
- Нейропептиды. Улучшают связь между нервными окончаниями.
Кроме медикаментов, иногда используют оперативное вмешательство для стимуляции блуждающего нерва. Для этого вводят генератор электроимпульсов.
Назначение такой операции возможно лишь в случае, если агенезия мозолистого тела грозит острыми патологиями важных органов.
Также операцию назначают, если другие методы терапии не дают никаких результатов. Установка генератора проводится в подключичную зону. После установки стимулятора ребенок должен посещать врача каждые несколько месяцев. Преимущество установки стимулятора при агенезии мозолистого тела – значительное сокращение количества приступов.
Пациенты лучше переносят любые симптомы, однако в некоторых ситуациях возможна неэффективность устройства. Иногда патология вызывает сколиоз, в этом случае для лечения назначают ЛФК и физиотерапию. Реже требуется операция на позвоночнике для улучшения состояния костных тканей.
Лечение и прогнозы
Агенезия правой почки или левой не представляет угрозы для жизни ребенка. Требуется динамическое наблюдение, соблюдение рекомендаций врача. Нужно беречь здоровый орган и предупреждать бактериальные заболевания мочевыводящей системы.
При отсутствии жалоб при аплазии левой почки или правой 1 раз в год проводится общий клинический осмотр. Прогноз благоприятный.
Питание должно быть сбалансированным, следует исключить жирные продукты. Термическая обработка – запекание, отваривание, тушение. Жареные и маринованные продукты следует исключить.
Алкоголь запрещен, так как этанол усиливает диурез и нагрузку на единственный орган.
При развитии компенсированной почечной недостаточности оформляется инвалидность. В зависимости от состояния пациента присваивается соответствующая группа.
Если почка справляется с нагрузкой, признаки ХПН отсутствуют, то инвалидность не оформляется. Термальная стадия почечной недостаточности – это показание для трансплантации органа, проводится диализ.
Двусторонняя агенезия несовместима с жизнью. Ребенок рождается мертвым либо погибает в первые сутки. Как правило, матери предлагают прервать вынашивание, вызвать искусственные роды.
Но современные методы диагностики позволяют выявить патологию в пренатальном периоде. После рождения показана трансплантация органа.
Пересадку проводят в первые сутки жизни ребенка. Далее пациенту необходим гемодиализ. Если не произошло отторжение органа, при нормальных показателях уродинамики процедуру внепочечной очистки крови отменяют.
При лечении заболеваний, не связанных с мочевыводящей системой, пациенту с трансплантатом или с односторонней агенезией должны назначаться препараты, не обладающие нефротоксическим действием.
Кроме этого, действующие компоненты лекарственных средств не должны выводиться с мочой.
Специфической профилактики при диагностированной агенезии почки не существует. Пациенту следует следить за питанием, избегать повышенных нагрузок на мочевыводящую систему.
Беременность возможна, но должна быть плановой и желанной. Аборты нежелательны. Наличие одной почки у матери не вызывает пороков развития у плода.
Женщина должна находиться под динамическим наблюдением у гинеколога. При отсутствии у женщины нарушений выводящей функции органа госпитализация в гинекологическое отделение не требуется.
Роды проводятся по решению гинеколога. Возможно как самостоятельное родоразрешение, так и кесарево сечение.
Прогноз при заболевании
Развитие частичной агенезии мозолистого тела в редких случаях приводит к патологиям, которые сокращают жизнь пациента. Подобная форма болезни возникает в 80% случаев и не сопровождается тяжелой симптоматикой. Изредка требуется неврологическая коррекция, ЛФК и физиотерапия.
Однако выраженные формы и средняя степень патологии провоцирует различные нарушения. В этом случае о благоприятном прогнозе говорить невозможно. Чаще всего основные нарушения – это проблемы с интеллектом, психическим развитием, выраженные неврологические патологии. Медикаментозная, народная терапия в этом случае не дает никакого эффекта.
Несмотря на встречающиеся благоприятные исходы, агенезия мозолистого тела относится к тяжелым неврологическим нарушениям, которые не поддаются успешной коррекции. Не удается установить ни точных причин, ни факторов, которые могли бы сократить число проявлений патологии.
Агенезия мозолистого тела – сложное заболевание головного мозга, поражающее человека только на стадии эмбрионального развития. Несмотря на это, обнаруживается она нечасто и во многих случаях не доставляет пациентам никаких проблем. Сроки обнаружения аномалии никак не влияют на успех ее лечения.