Without health, happiness is impossible Vissarion Belinsky
Especially when it comes to our long-awaited babies. All mothers carefully examine their treasure during daily care, and any change in the baby’s appearance causes great concern. The greatest fear appears when we see the presence of problems associated with the development of our child’s head and brain.
But know that any diagnosis made by doctors is not a death sentence! When the baby is still very small, all violations can be corrected with the right approach. The more we know, the more effective our actions will be.
general information
The skull of a newborn baby consists of six separate bones: two parietals, two temporal, occipital and frontal. Between them lie thin layers of fibrous elastic tissue - cranial sutures. The areas where several bones of the skull connect are called fontanelles or “soft spots.”
Sutures are necessary so that the skull bones can partially overlap during passage through the birth canal. This makes the birth process easier and reduces the risk of brain damage to the newborn. Closing of the fontanelles occurs during the first year of a child’s life.
In 1 out of 1000 children, intrauterine or early fusion of cranial sutures is detected - craniostenosis. Pathology is more common in boys. There are three forms:
- monosynostosis (the most common type) - one suture is closed - metopic, sagittal, coronal or lambdoid;
- polynostosis – more than two sutures are closed,
- pansynostosis – more than three sutures are missing.
Craniostenosis in newborns can be an isolated anomaly or be part of a syndrome (20% of cases) and be combined with other developmental defects.
Prevention
During pregnancy, smoking and a hyperphosphate diet should be completely avoided (artificial drinks like cola and others, sausages, artificial foods with large amounts of phosphorus-containing leavening agents, humectants and other additives that give products a considerable shelf life and “high-quality” appearance). Balanced nutrient support should be used in combination with a comprehensive intake of vitamins and minerals in a synergistic combination and physiological dosages.
Share:
Facebook Youtube Viber Telegram
Causes
The reasons for the development of complete or partial craniostenosis have not been reliably established. The development of pathology is based on a disruption in the formation of bone tissue of the skull, which begins at the end of the first month of intrauterine development. It is assumed that the failure is caused by one of the following factors:
- hormonal imbalance;
- intrauterine infections (flu, herpes, rubella);
- genetic pathologies (defect in the gene encoding fibroblast growth factor receptors);
- compression of the fetal head in the uterus.
Normally, by the fifth month of pregnancy, the fetal skull bones should be well formed and separated by sutures. If craniostenosis occurs, brain function is disrupted and ophthalmological pathologies develop.
Craniosynostosis
Craniosynostosis
is a disease the main symptom of which is deformation of the cerebral part of the skull, resulting from premature healing of bone sutures.
The clinical picture includes skull deformations, symptoms of intracranial hypertension, pathology of the optic nerve, and mental retardation. Rarely, the disease is accompanied by abnormalities of the bones of the facial skull.
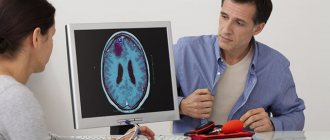
Diagnosis consists of assessing the degree of fusion of cranial sutures and identifying bone defects through physical examination, radiography, CT and MRI. The main treatment is early surgical correction of the shape of the skull bones.
Craniosynostosis is a pathological condition in pediatrics that occurs against the background of early fusion of cranial sutures, characterized by deformation of the skull and impaired development of brain tissue.
On average, the prevalence of various forms of the disease in the CIS countries is 0.03-3.5% of all newborns. The male sex is more prone to developing this pathology. The most common variant is monosynostosis.
Most often, premature overgrowth of the sagittal suture (scaphocephaly) is observed - 50-65% of all craniosynostoses. The rarest and most prognostically unfavorable is the syndromic form, in which there is a high risk of death in the first year of a child’s life.
With timely diagnosis and adequate treatment in the first 6-9 months of life, the patient’s further development proceeds without deviations.
Craniosynostosis
This pathology also occurs with hereditary pathologies - Apert syndrome, Crouzon syndrome and Pfeiffer syndrome.
One of the leading causes of the development of craniosynostosis is known for certain - an anomaly of the gene responsible for the formation of fibroblast growth factor receptors (FGFR types I, II and III).
Pathogenetically, craniosynostosis is caused by premature synostosis of one or several cranial sutures: coronal, sagittal, lambdoid or metopic. Against this background, according to Virchow's law, compensatory growth of bone tissue occurs in the perpendicular direction, which causes deformation of the skull.
Polysynostosis (and often monosynostosis) is often accompanied by intracranial hypertension, which can manifest itself as neurological disorders due to compression of the cerebral cortex, venous stagnation of the fundus, swelling of the optic disc, and in the long-term course - complete atrophy of the optic nerve and loss of vision.
Classification of craniosynostosis
Craniosynostosis, according to etiological factors, is divided into two groups:
- Syndromic. In this case, the pathology is combined with other congenital defects. These include X-linked, monogenic, chromosomal and other craniosynostoses. For example, a combination of synostosis with dysplasia of the facial bones, Smith-Lemli-Opitz syndrome or oro-finger-facial syndrome.
- Non-syndromic. This is an isolated form that occurs independently and has no associated diseases.
Depending on the number of overgrown cranial sutures, the following are distinguished:
- Monosynostosis. Characterized by damage to only 1 suture. In the case of the coronal and lambdoid suture, the overgrowth can be one- or two-sided. The most common form.
- Polysynostosis. 2-3 sutures are involved in the pathological process.
- Pansynostosis. With this form, fusion of all the bone sutures of the child’s skull is observed. It is extremely rare.
Clinically, craniosynostosis manifests itself from the moment the child is born. All forms are characterized by plagiocephaly and early closure of the large fontanel (normally this occurs at 12-18 months).
Only with polysynostosis or concomitant hydrocephalus can it remain open until 3 years of age.
Also, with craniosynostosis, an increase in intracranial pressure is often observed, which can be manifested by neurological disorders: anxiety, intense crying, nausea and vomiting, sleep disturbance, loss of appetite, positive Graefe's symptom, convulsions.
Each form of the disease has characteristic clinical features. Craniosynostosis of the sagittal suture (scaphocephaly or scaphoid skull) is characterized by an increase in the anteroposterior size of the child’s head with insufficient width.
The elongation of the skull, “indentation” of the temporal regions, “overhanging” of the forehead and occipital part, narrowing of the face and its acquisition of an oval shape are visually determined. Palpation reveals a bone ridge above the passage of the sagittal suture.
At an early age, mental development may be delayed.
Overgrowth of the lambdoid suture is most often unilateral and manifests itself as flattening of the occipital region. It is a difficult form to diagnose, since plagiocephaly is almost invisible under the hair, and neurological disorders are minimal. As the patient grows older, the dynamics of the disease are practically absent.
Over time, a curvature of the nose in the opposite direction, flattening of the cheekbones, malocclusion and strabismus develop. Bilateral coronary craniosynostosis is manifested by a wide, flat and high forehead with flattened orbital margins of the frontal bone, and rarely by tower deformation of the skull (acrocephaly).
Neurological disorders are nonspecific and similar to other forms.
Nontopic craniosynostosis or trigonocephaly is characterized by the development of a triangular forehead with a bony keel extending from the glabella to the greater fontanel.
Hypotelorism is also observed - posterior displacement of the orbits with a decrease in the interorbital space. Over time, there is some smoothing of the bone ridge and normalization of the shape of the forehead.
In half of the cases, visual impairment and mental retardation occur.
Syndromic craniosynostosis is the rarest and most severe form. In addition to plagiocephaly, there is dysplasia of the bones of the facial part of the skull, which causes respiratory failure, eating disorders and vision pathology. It is characterized by synostosis of the coronal suture and, as a result, a brachycephalic shape of the child’s head.
Hypoplasia of the bones of the upper jaw, protrusion of the eyeballs from the orbits, and hypertelorism also occur. Often there is a significant expansion of the fontanel and divergence of the sagittal suture.
Without treatment, children develop severe mental retardation and often die during the first 12 months of life from acute respiratory viral infections complicated by pneumonia.
An important point is a visual examination of the child, which makes it possible to detect characteristic deformations of the skull, bone anomalies, etc.
Laboratory tests do not reveal specific changes and can be used to determine genetic pathology or diagnose complications.
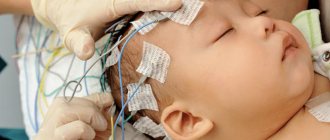
Instrumental methods are required to visualize bone deformations and assess the degree of damage to brain tissue. This includes neurosonography, radiography, computed tomography and magnetic resonance imaging.
Neurosonography is used to assess the condition of brain tissue and the size of the ventricles, and to identify intracranial hypertension.
An x-ray can detect abnormalities in bone structure, ossification of cranial sutures, and, with increased intracranial pressure, increased digital impressions. CT and MRI are used to obtain more informative results.
If damage to the visual system is suspected, ophthalmoscopy is performed to detect damage to the optic nerve head. Consultations with a neurosurgeon and ophthalmologist are recommended.
Differential diagnosis of craniosynostosis is carried out with positional plagiocephaly, birth trauma of newborns (cephalohematoma, subgaleal hemorrhage, skull fracture), brain cysts, rickets and microcephaly.
The main treatment for craniosynostosis is surgical correction of the bony deformity of the skull. The optimal time for surgical intervention is the first 6-9 months of a child’s life.
These periods are due to the fact that during this period the most intensive development of brain tissue is observed, which can be hampered by deformation of the cranium. In addition, the bones of the skull at this age quickly restore their structure without the development of complications.
In addition to surgical treatment, the child’s diet is changed in accordance with age requirements. With the development of intercurrent diseases, drug therapy is indicated.
Forecast and prevention of craniosynostosis
The prognosis for children with craniosynostosis directly depends on the form of the disease, the timeliness of diagnosis and the effectiveness of surgical intervention. With high-quality treatment, the outcome of the disease is usually favorable. The syndromic form of craniosynostosis is considered to be unfavorable prognostically.
There is no specific prevention for this pathology. Nonspecific measures include medical and genetic consultation of the family and pregnancy planning, protecting the woman’s health while bearing a child, balanced nutrition, giving up bad habits and excluding all potential etiological factors for the development of craniosynostosis.
Source: https://www.KrasotaiMedicina.ru/diseases/children/craniosynostosis
Symptoms
The severity of symptoms of craniostenosis depends on how many sutures are closed and during what period it occurred. With intrauterine growth, the clinical picture is more pronounced.
The main external manifestation of the pathology is skull deformation, which occurs due to compensatory bone growth perpendicular to the affected suture (Vikhrov’s law). The characteristics of the deformation are determined by which seams closed prematurely. There are three types of pathology:
- scaphocephaly - overgrowth of the sagittal suture, which divides the head in the parietal part into the right and left halves;
- plagiocephaly - closure of the coronal suture, which runs perpendicular to the sagittal one from ear to ear;
- trigonocephaly - closure of the metopic suture dividing the forehead into two equal parts;
- brachycephaly - fusion of the lambdoid and coronal sutures.
Sagittal craniostenosis (scaphocephaly) is the most common type of disease. The width of the skull is reduced, its length in the longitudinal (antero-posterior) direction is increased. The occipital and frontal bones overhang. The temples seem depressed. The child's face has a narrow oval (scaphoid) shape. Neurological symptoms are not typical for scaphocephaly.
Plagiocephaly can be frontal (frontal) or occipital, and unilateral or bilateral. The most common finding is unilateral frontal coronary craniostenosis. In this case, the skull is deformed asymmetrically. On one side, the forehead thickens, the eye socket and eyebrow rise, the auricle protrudes, the nose bends, the cheekbone flattens, the jaw shifts, and so on. Morphological changes occur as the child grows older. Strabismus develops.
Brachycephaly is characterized by an increase in the transverse diameter of the skull and a decrease in its height. Supplemented by ophthalmological and neurological signs.
Trigonocephaly is accompanied by a wedge-shaped protrusion of the forehead forward - a bone keel is formed, running from the large fontanelle to the glabella. The deformity is combined with hypotellorism and visual impairment.
Other possible signs of craniostenosis (usually appear after 12 months):
- hydrocephalus;
- intracranial hypertension;
- headache;
- nausea, vomiting;
- convulsions;
- protruding eyes (exophthalmos);
- delayed intellectual development;
- meningeal symptoms.
Craniosynostosis in children
Craniosynostosis (or craniostenosis) is a birth defect in which one or more cranial sutures close before the baby's brain is fully formed.
Typically, the fontanelles close by the age of two, forming a solid bone. The fontanelles, which do not ossify until a certain point, provide the child’s brain with sufficient space for normal growth. If the sutures heal too early, the growing brain rests against the skull, which limits its growth. As a result, the child develops various deformations of the skull. Craniosynostosis leads to increased intracranial pressure, which in turn leads to vision loss, mental retardation and other complications.
Types of Craniosynostosis
There are different types of craniosynostosis. They are distinguished by the nature of the deformation of the skull, depending on which sutures are affected by the pathology, and the causes of the disease. In 80-90% of cases, only one suture is involved in the pathology.
There are two main types of craniosynostosis.
The non-syndromic type is the most common. It is believed to be caused by a combination of genetic and environmental factors. Syndromic craniosynostosis is usually caused by inherited syndromes such as Apert syndrome (acrocephalosyndactyly), Crouzon syndrome (craniofacial dysplasia), and Pfeiffer syndrome.
Depending on the number of overgrown sutures, they distinguish between monosynostosis - pathology of one suture, polysynostosis - several, and pansynostosis - fusion of all cranial sutures.
Forms of craniosynostosis can also be classified according to the affected suture:
Scaphocephaly - fusion of the sagittal suture
The most common type. The sagittal suture in the upper part of the cranial vault is affected. As the baby grows, the baby's head becomes abnormally narrow and long.
Frontal (fusion of the coronal suture) or occipital (fusion of the lambdoid suture) plagiocephaly
In this case, unilateral fusion of the coronal or lambdoid suture occurs.
With frontal plagiocephaly, the forehead is flattened on the side of the synostosis, and a compensatory bulge is formed on the opposite side.
If the sutures close on both sides - brachycephaly - the baby's head will be shorter and wider than usual. In especially severe cases, the head takes on a “tower” shape – acrocephaly.
Trigonocephaly - fusion of the metopic (frontal) suture
With this type, pathology occurs in the metopic (frontal) suture, which runs from the top of the cranial vault to the bridge of the nose. In children with this type of craniosynostosis, the head takes on a triangular shape, a bony ridge forms at the site of the suture, and the eyes are very close-set.
Symptoms of craniosynostosis
Symptoms of craniosynostosis usually appear immediately after birth or several months later. The symptoms are as follows:
- skull deformation
- abnormal appearance of the fontanel or its absence in the upper part of the calvarium
- formation of a bone ridge at the site of fusion of a suture that closed too early
- abnormal growth of a child's head
Depending on the type of craniosynostosis, your child may have other symptoms:
- headache
- close or wide set eyes
- reduced learning ability
- vision loss
Doctors diagnose craniosynostosis during a physical examination. Sometimes a CT scan is used, which reveals the closure of the sutures in the child's skull. Genetic tests and physical characteristics help the doctor determine the presence of syndromes that contribute to the development of this disease.
Causes of craniosynostosis
1 child in 2500 is born with this disease. It is more common in boys. In most cases, the disease develops accidentally, but in a small number of children with this pathology, the skull bones fuse too early due to genetic syndromes. Such as:
- Apert syndrome
- Carpenter's syndrome
- Crouzon syndrome
- Pfeiffer syndrome
- Saethre-Hotzen syndrome
Treatment of craniosynostosis
A small number of children with mild craniosynostosis do not require surgical treatment. It is enough for them to wear a special corrective orthosis that fixes the shape of the skull while their brain develops.
But most children with this pathology require surgery to correct the shape of the skull and reduce intracranial pressure.
What kind of operation will be performed depends on which sutures are affected and what exactly was the reason for the development of craniosynostosis.
Surgeons correct affected sutures in the following ways:
Endoscopic surgery
Endoscopy is best suited for infants under 3 months, but can also be used in children under 6 months if only 1 suture is closed.
During this procedure, the surgeon makes 1-2 small incisions on the child's head. A thin light tube with a camera at the end is inserted into the cranial cavity, and a small strip of bone is cut out with manipulators at the site of the overgrown suture.
With endoscopic surgery, the risk of massive blood loss is reduced and the recovery period is faster. After surgery, the child may need to wear a corrective orthosis for about a year to change the shape of the skull.
Open surgery
Open surgery is performed on children under 11 months of age.
The surgeon makes one large incision in the child's skull. Removes bones from the affected area and changes their curvature. The reconstructed bones are secured to other bones with absorbable plates and screws. Some children require more than one surgery to correct the shape of their skull.
Children who have had this surgery do not need to wear a brace. However, open surgery is accompanied by large blood loss and a longer recovery period than endoscopic surgery.
Types of surgical treatment of craniosynostosis
Surgical treatment of craniosynostosis depends on the sutures involved, the age of the child and his individual needs.
The most commonly recommended treatment options include:
Linear craniotomy
Treatment includes traction of the bones of the cranial vault and a procedure to change the curvature of bone fragments. Linear craniectomy may also be performed as a preliminary procedure to relieve intracranial pressure in very young children (usually <6 months) in whom multiple sutures have been affected by pathology.
Typically, linear craniectomy is performed jointly with a pediatric neurosurgeon. The procedure usually takes approximately 2-3 hours. After the operation, the child remains in the hospital for recovery and rehabilitation. Most children stay in the hospital for an average of three to five days.
Additional operations, including traction of the bones of the cranial vault and restoration of the natural shape of the skull, are prescribed depending on the speed of the child’s recovery and response to primary treatment.
Sagittal spring distraction devices
The surgery involves a linear craniectomy and the placement of two or three stainless steel springs into the gap between the bones to increase the distance between them and create additional space for brain development, to improve the shape of the skull and reduce the risk of sagittal suture reclosure.
Such springs are an effective “minimally invasive” method that does not require a long hospital stay.
The spring devices are placed through small incisions in the skull, limiting blood loss so much that blood transfusions are required in a minimum number of patients. After about three months, additional surgery is needed to remove the springs.
A cranial orthosis can be used before surgery to reduce the progression of calvarial deformation that is inevitable with sagittal synostosis, as well as after surgery to remove spring devices to correct the process of straightening the shape of the skull.
Fronto-orbital reduction
Fronto-orbital reduction is used to correct metopic or coronal synostosis and for simultaneous fusion of multiple sutures. It involves exposure and sawing out of the supraorbital block and a fragment of the frontal bone through an ear to ear incision.
After which the frontal bone is removed. The supraorbital block is remodeled (advanced until symmetry with the unaffected side is achieved) and fixed in the correct position with absorbable plates and screws. Then the bend of the frontal bone is formed, which is closest to the physiological curvature, and it is fixed to the supraorbital block.
This procedure provides protection for the eyes, creates additional volume in the cranial cavity for further brain development, and remodels the supraorbital block and frontal bone to form a normal forehead convexity.
Fronto-orbital reconstructive surgery usually takes two to five hours, with an average postoperative hospital stay of four to five days for observation and recovery. As the child grows and develops, he may require additional manipulations.
Remodeling and reconstruction of the parieto-occipital region
Remodeling or reconstruction of the bones of the cranial vault is a one-stage operation that involves changing the shape of the bones. This operation is used if more than one suture is stenotic.
After the bone is removed, it is remodeled and stretched, for example, with a bilateral occipital craniotomy, the sawed fragment of the occipital bone is remodeled by creating radial notches on the bone, thus giving it the necessary curvature until symmetry is achieved.
The remodeled skull is strengthened with bone graft and absorbable plates or sutures to give the brain more room to develop and the head to be shaped as closely as possible. This operation requires hospitalization for several days. A postoperative drainage tube is also placed and removed before leaving the hospital.
What is important: after a total reconstruction of the bones of the cranial vault, children are not required to wear a cranial modeling orthosis, and when they grow up they will be able to play sports.
Distraction (traction) of the occipital region of the cranial vault. Distraction-compression devices.
Distraction-compression devices are used in neurosurgery and are intended to treat patients with craniocerebral disproportions (mainly children), by gradually stretching the bones of the occipital region in order to increase intracranial space.
The traction technique today often replaces traditional fronto-orbital reconstruction, which is still the most common primary treatment for patients with syndromic craniosynostoses. The method of distraction of the occipital region of the cranial vault has a number of advantages.
Distraction devices stretch the soft tissue along with the bone, thereby increasing the distance of traction. This is especially useful if your child has increased intracranial pressure or severe head deformity.
During the procedure, a coronal incision is made (from ear to ear), the back of the arch is exposed and the bone of the desired shape is cut out for subsequent stretching. A distraction device is installed in the gap between the bones and rigidly fixed with microscrews, to the fixed part of the skull and to the retractable part, then the incision is closed. Traction machines work by very slowly stretching the bone and surrounding tissue.
The operation takes approximately two to three hours. The post-operative recovery period will take two to three days. This operation is less invasive than traditional open calvarial traction surgery and provides greater traction for both bones and soft tissues.
A second surgery will be required in approximately three months to remove the distractors.
Final facial contouring for craniostenosis
As your child matures and all major osteotomies are behind them, they will require final facial contouring to correct any remaining facial irregularities and improve their overall appearance.
This procedure involves smoothing out irregularities and restoring the bones of the facial part of the skull, installing bone grafts or bone substitutes if necessary, and restoring normal facial proportions.
Complications of craniostenosis
Surgery will help prevent complications that usually accompany craniosynostosis. If no action is taken, the shape of the baby's head will eventually change irreversibly.
As a child's brain grows, the pressure inside the skull constantly increases, which will later cause problems such as blindness and mental retardation.
Complications that may occur during surgery:
- blood loss (caused by damage to the venous sinuses)
- damage to the dura mater accompanied by brain damage
- abnormalities of bone tissue (bone destruction or inadequate regeneration)
- recurrence of craniosynostosis and the appearance of new deformities.
Prognosis (prospect) for craniostenosis
A successful operation allows the child's brain to develop normally. Most children who undergo surgery have a normal skull shape and do not have developmental delays or other complications.
Where in Russia is surgery performed for craniosynostosis?
If you are looking for a clinic where you can have surgery for craniosynostosis, there are good reviews on the Internet about the surgeon Sergei Yasonov, who operates at the Russian Children's Clinical Hospital in Moscow. On his page you can see examples of photographs of children with craniosynostosis before and after surgery.
Link instagram.com/dr.sergey_yasonov/
Maria Shekhter
Source: https://carence.ru/2018/08/kraniosinostoz-u-detej/
Diagnostics
Craniostenosis is diagnosed based on:
- collecting anamnesis and analyzing symptoms;
- visual examination of the patient’s skull and craniometry (measurement of its parameters using a tape);
- palpation of the fontanelles;
- neurological and ophthalmological examinations;
- fundus examination to assess intracranial pressure;
- radiography;
- CT or MRI of the brain.
These methods make it possible to detect the absence of cranial sutures, intracranial hypertension, ophthalmological pathologies, changes in the structure of the brain and other disorders.
Craniostenosis is differentiated from cranial deformations that are not due to synostosis of the sutures. In addition, the purpose of diagnosis is to determine concomitant developmental defects of the child.
Complications
In addition to external changes, the disease can lead to big problems in the normal development of a little person and the appearance of many concomitant diseases:
- Pathology of the structure of the entire skeleton (vertebral fusion).
- Improper development of joints, which leads to disability.
- Difficulty in nasal breathing (night apnea, in which the baby temporarily stops breathing).
- Hydrocephalus.
- Retarded physical and psychological development.
- Strabismus, blindness, tremor.
- Underdevelopment of the jaws and eye sockets.
Craniosynostosis leads to such severe complications in extreme forms, when fusion of several areas of the skull occurs. There are several degrees of the disease, which depend on the number and type of closed sutures.
Treatment
How is craniostenosis treated in a child? The main method of therapy is surgery. Its goal is to enlarge the cranial cavity and give it an anatomical shape by cutting fused sutures or remodeling the bones of the vault using distraction devices.
It is advisable to perform surgery for craniostenosis at 6-9 months. Up to 2 years of age, the brain is actively growing. Therefore, intervention at an early age allows for regression of all pathological manifestations, including elimination of deformation of the cranial and facial bones. The effectiveness of surgical treatment of craniostenosis is evidenced by photographs after surgery.
Child with craniosynostosis before and after surgery
After 2-3 years, surgery has virtually no effect on the functions of the brain and visual analyzer. Its result is an improvement in the child's appearance.
It is possible to do without surgery for craniostenosis if isolated scaphocephaly without neurological disorders is observed.
Etiology and pathogenesis
The reasons for premature suture closure are still not fully understood. The most common factors for the development of craniostenosis are:
- Intrauterine pathology;
- Hormonal imbalance;
- Genetic disorders;
- Mechanical impact on the head.
However, in most cases of craniostenosis, it is not possible to identify the root cause of its occurrence.
Craniosynostosis leads to the formation of insufficient space in the cranial cavity, which in turn mechanically compresses the actively growing brain. Such craniocerebral disproportions in a child of the first year of life are considered as causes of blood circulation disorders. On the one hand, venous outflow is disrupted. As a result, an uncontrolled increase in intracranial pressure occurs. On the other hand, symptoms of increasing hypoxia form multiple foci of ischemia in the brain.
Forecast
The consequences of craniostenosis depend on the severity of the deformities and the number of affected sutures. The isolated form of the pathology has a favorable prognosis subject to surgical intervention. Without surgery, craniostenosis in adults leads to complications such as:
- persistent pronounced change in the shape of the skull;
- headache;
- periodic seizures;
- strabismus.
Syndromic craniostenosis is usually associated with disability.
Classification
Scaphocephaly is classified according to its clinical picture. There are two types of pathological process:
- a syndrome in which there is a significant protrusion of the skull in the frontal region;
- a syndrome in which the frontal part is normal, but the occipital part is much larger than normal for age.
The impact of both the first and second forms of pathology, if correct treatment is not started in a timely manner, is extremely negative: the consequence may be irreversible mental retardation. Scaphocephaly is one of the most latent forms of craniostenosis, since cognitive impairment occurs only in the absence of therapy.
What diseases can be confused with craniosynostosis?
Postural deformation of the skull
- children with motor deficits;
- rickets (soft bones of the skull);
- osteogenesis imperfecta
Secondary causes
- microcephaly;
— excessively rapid decompression of hydrocephalus (cerebrospinal fluid pressure decreases below normal);
- primary hyperthyroidism;
- overdose in the treatment of hypothyroidism;
- hypophosphatasia;
- vitamin D-resistant rickets;
- mucopolysaccharidosis;
- osteopetrosis.